
2006 год
Главная иллюзия медицины — иллюзия формальной логики.
Нормальная микрофлора и дисбактериоз
- Нормальная микрофлора: общие принципы и частности.
- Добро и зло естественных микробиоценозов.
- Дисбактериоз как микробиологическое понятие.
- Дисбактериоз и патология: причина или следствие?
- Дисбактериоз как индикатор здоровья.
- Пробиотики и фаготерапия: святая простота формальной логики.
Органы человека, контактирующие с внешней средой, уже в момент рождения инфицируются различными микробами. Большинство из них проходит транзитом, другие задерживаются на непродолжительное время. Но есть и такие, которые, находя благоприятные для себя условия, размножаются, создавая то, что называется нормальной микрофлорой. Количество бактерий, населяющих покровные ткани (кожу, слизистые оболочки), во много раз превосходит число собственных клеток хозяина.
Микробы кишечника представляют доминирующий в количественном и качественном отношениях компонент нормальной микрофлоры человека. Главным полигоном является толстый кишечник — коллектор пищевых отходов, великолепного питательного материала для бактерий, грибов, простейших. Вкупе с физическими (температура, влажность, содержание кислорода) и химическими (рН) параметрами это создает благоприятные условия для развития многочисленных микроорганизмов. Лидерство принадлежит бактериям. Пользуясь благами симбиоза, они составляют базис нормальной (физиологической) микрофлоры, которая сопутствует своему хозяину на протяжении всей его жизни.
Вопрос о плате бактерий за предоставляемые удобства дискутировался со времен Л. Пастера, который рассматривал нормофлору как абсолютно полезный и необходимый атрибут. Альтернативой была концепция интестинальной интоксикации. Она стращала ядами гнилостных бактерий, опасных для организма. На биохимическом языке гниение означает ферментацию белков (брожение — ферментация углеводов), наиболее общим итогом которой является аммонификация, т.е. минерализация органического азота. Этот необходимый для биосферы процесс сопровождается высвобождением ядовитых веществ типа индолов, скатола, кадаверина, путресцина, гистамина и пр. В кишечнике гниение связано главным образом с анаэробами и энтеробактериями.
Концепция об интестинальной интоксикации нашла активного пропагандиста в лице И.И. Мечникова. Он рассматривал ее как длительный, хронический процесс, который, продолжаясь в малых дозах всю жизнь, вносит существенный вклад в развитие болезней и старение организма. Позитивная функция нормальной микрофлоры, по его мнению, состоит лишь в сопротивлении, которое она оказывает гнилостным бактериям толстого кишечника. Такую роль он отводил бактериям, сбраживающим углеводы до молочной кислоты. Кислая среда препятствует размножению гнилостных бактерий подобно тому, как это происходит при скисании молока или при квашении овощей. Иными словами, по И.И. Мечникову, «доброе начало» нормальной микрофлоры сводится исключительно к регуляции собственного гомеостаза, а потому уничтожение всей микрофлоры не должно иметь отрицательных последствий для хозяина. В своих рекомендациях он доходил до крайностей, таких как целесообразность резекции толстого кишечника и замещения его дикой микрофлоры культурными бактериями.
Эта идея стала основой для стремления радикально изменить микробиоценоз кишечника, превратив его в благоухающий резервуар молочнокислых бактерий. Но все попытки добиться столь заманчивого результата провалились. Этого следовало ожидать, так как претензии И.И. Мечникова противоречили эволюции, закрепившей за каждым видом животных определенный тип микробиоценоза. Кстати, и само гниение — собирательный, многоэтапный процесс, который реализуется благодаря совместной деятельности многих бактерий. Большинство из них участвует и в бродильных процессах, вырабатывая различные органические кислоты, в том числе молочную кислоту. Все зависит от того, в какие условия они поставлены, а эти условия создает прежде всего сам хозяин. На нормальную микрофлору следует смотреть как на эволюционно закрепившуюся неизбежность. Ее необходимо правильно понимать, а управлять ей требуется лишь в тех случаях, когда мы сами нарушаем гомеостаз этого сложного, многофакторного сообщества, которое живет по собственным законам, опираясь на физиологию хозяина.
Количественные и качественные нарушения нормальной микрофлоры принято называть дисбактериозом. Развитием этого понятия является дисбиоз — термин, который отражает стремление охватить изменения не только среди бактерий, но и среди других представителей нормофлоры. Фактически это декларация, так как в современных анализах на дисбиоз учитывается единственный небактериальный компонент — дрожжеподобные грибы рода Candida. Заметим также, что понятия дисбактериоз/дисбиоз используются только в отечественной литературе.
Проблему дисбактериоза чаще всего анализируют на примере микрофлоры толстого кишечника, так как она наиболее разнообразна, обильна и легкодоступна для исследования. По сути вся концепция о дисбактериозе построена на изучении микрофлоры фекалий, являясь частью копрологии. При этом предполагается, что количественная и качественная необычность микробного пейзажа испражнений таит в себе опасность, требуя коррекции. Иными словами, бактериологический диагноз «дисбактериоз» трансформируется в клиническое понятие с вытекающими отсюда последствиями для носителя такого диагноза. Такая идеология прочно укрепилась в сознании врачей, и дисбактериоз стал одним из самых популярных диагнозов, особенно в педиатрии. Его ставят, начиная с рождения ребенка, при первых нарушениях стула, симптомах диатеза, аллергии, при кожных заболеваниях, инфекционной гипорезистентности и пр. Подобная практика, не имея аналогов за рубежом, проводится в нашей стране не одно десятилетие. За это время представления о нормальной микрофлоре претерпели серьезную эволюцию. Утверждено количественное и функциональное лидерство за облигатными анаэробами, определены экологические, клинические и ятрогенные ситуации, способствующие дестабилизации микрофлоры, сформулированы понятия о колонизации и колонизационной резистентности, возникло учение об оппортунистических инфекциях. Все это побуждает пересмотреть патогенетические аспекты проблемы дисбактериоза/дисбиоза, оценив их реальные позиции в современной медицине.
Двенадцатиперстная кишка и проксимальный отдел тощей кишки практически стерильны. По мере приближения к подвздошной кишке число бактерий увеличивается, достигая 105 в 1 мл кишечного содержимого (преимущественно лактобациллы и энтерококки). Подвздошная кишка богаче микрофлорой, разнообразие которой нарастает к дистальным отделам, где примерно поровну представлены анаэробные (бактероиды, бифидобактерии и др.) и факультативно анаэробные бактерии (кишечная палочка, лактобациллы, энтерококки).
После баугиниевой заслонки (т.е. с того места, где начинается толстый кишечник) картина резко меняется. Общее количество жизнеспособных бактерий в 1 г фекалий возрастает до 1010—1012, а качественный состав усложняется до 400 и более видов (см. таблицу). Абсолютное большинство составляют бесспоровые анаэробы, среди которых доминируют бактероиды, бифидобактерии, эубактерии, катенобактерии, пептококки. Часто присутствуют спороносные анаэробы (клостридии), количество которых, впрочем, значительно уступает бесспоровым формам. На долю аэробной флоры приходится всего 1—5%; она распределяется примерно поровну между энтеробактериями (преобладает кишечная палочка), энтерококками и лактобациллами. Встречаются также стафилококки и дрожжеподобные грибы (кандиды); их количество не превышает 104—106/г фекалий.
Нормальная микрофлора желудочно-кишечного тракта взрослого человека
| Микроорганизмы | Концентрация бактерий (в 1 мл/г) | |||
| Желудок | Тощая кишка | Подвздошная кишка | Толстый кишечник | |
| Общее число жизнеспособных бактерий | 0—103 | 0—105 | 102—107 | 1010—1012 |
| Аэробы и факультативные анаэробы: | ||||
| энтеробактерии | 0—102 | 0—103 | 102—107 | 104—1010 |
| энтерококки | 0—102 | 0—104 | 102—106 | 105—1010 |
| стафилококки | 0—102 | 0—103 | 102—105 | 104—109 |
| лактобациллы | 0—103 | 0—104 | 102—105 | 104—1010 |
| грибы (кандиды) | 0—102 | 0—102 | 102—104 | 104—106 |
| Анаэробы:бактероиды | Редко | 0—103 | 103—107 | 1010—1012 |
| бифидобактерии | Редко | 0—104 | 103—109 | 108—1012 |
| пептококки | Редко | 0—103 | 102—106 | 1010—1012 |
| клостридии | Редко | Редко | 102—106 | 105—1011 |
| эубактерии | Редко | Редко | Редко | 109—1012 |
Цифры, приводимые разными авторами, значительно варьируют. Кроме технических причин, это связано с колебаниями индивидуальных показателей, включая данные, полученные у одних и тех же лиц в разные сроки обследования. Это делает понятие нормальной микрофлоры скорее концептуальным, чем формальным, вынуждая допускать такие вольности, которые неприемлемы для других показателей. Чтобы избежать противоречий, предложено дифференцировать резидентную (син. индигенная, аутохтонная) и факультативную флору, подразумевая облигатный характер первой из них и случайный, временный — второй. Основные особенности микрофлоры связаны с ее факультативным компонентом. Кроме того, фекальную флору нельзя считать полноценным отражением микробного пейзажа слизистой оболочки толстого кишечника: она характеризует скорее полостную, чем пристеночную микрофлору. Последняя более стабильна и физиологична.
Механизмы формирования нормальной микрофлоры
Стерильность плода исчезает при прохождении родового канала. Едва вступив в мир, новорожденный сталкивается с множеством бактерий, всасывая их с материнским молоком, заражаясь при контакте со взрослыми и окружающей средой. Кишечная микрофлора младенца имеет ряд особенностей, которые определяются своеобразием вскармливания. Лактоза (которой богато молоко) особенно активно метаболизируется лактобациллами и бифидобактериями. Кроме того, женское молоко содержит N-ацетилглюкозамин, стимулирующий рост бифидобактерий; его нет в коровьем молоке. Это способствует опережающему размножению бифидобактерий и их повышенному содержанию в кишечнике новорожденных при естественном вскармливании. Количество других бактерий ниже, чем у взрослых и у новорожденных, лишенных грудного молока. Это объясняется неблагоприятным для многих бактерий закислением среды активно растущими бифидобактериями (результат сбраживания лактозы). Впрочем, уже в этом периоде микрофлора содержит практически все зачатки будущего микробиоценоза и с завершением грудного кормления быстро обретает «взрослые» характеристики.
Способность бактерий к колонизации слизистых оболочек определяется двумя главными механизмами — адгезией (закреплением) на поверхности эпителиоцитов и выживанием в новом окружении. Нормальные обитатели слизистых оболочек и патогенные микроорганизмы в равной мере обязаны преодолеть ряд препятствий, чтобы завоевать право на колонизацию. Однонаправленный поток материала через большинство висцеральных каналов (результат перистальтических движений, гравитационных сил или мукоцилиарного транспорта) угрожает выталкиванием и/или вымыванием микробов. Для того, чтобы удержаться (например, в пищеварительном канале), они должны зацепиться за эпителиальную поверхность. Прикрепление к эпителиальным клеткам не только предотвращает элиминацию микробов, но и стимулирует их рост из-за повышенной концентрации питательных веществ на границе твердой и жидкой фаз.
Адгезия бактерий отличается специфичностью. Например, лактобациллы птиц прикрепляются только к птичьим эпителиоцитам, тогда как лактобациллы крысы предпочитают крысиный эпителий. Можно полагать, что избирательное сродство микробов к эпителиальным клеткам детерминирует их селективный симбиоз с определенными видами животных. Еще удивительнее то, что бактерии способны выбирать типы эпителиальных клеток, закрепляясь на тех из них, к которым имеют больше сродства. Это наблюдается даже в пределах одной и той же анатомической области, например в ротоглотке и ротовой полости. Адгезивные свойства меняются при культивировании бактерий, и уже через несколько инвитровых пассажей штамм может потерять способность к колонизации эпителиоцитов. Заметим и то, что эпителиоциты покрыты слоем слизи, который должны преодолеть бактерии, чтобы добраться до поверхности клетки. Не всем это удается, и такие бактерии обречены на быстрое удаление вместе со слизью.
Тканевой тропизм определяется структурными особенностями внешних компонентов (адгезинов) бактерий, которые избирательно связываются с рецепторами клеток. Специфичность многих адгезинов определяется их принадлежностью к лектинам (лат. legdo, выбирать) — белкам, которые комплементарны определенным углеводным радикалам, неодинаково представленным на разных типах клеток. Возможен и обратный вариант: лектиноподобные рецепторы клеток распознают и связываются с углеводами бактерий. Кроме того, бактерии закрепляются на фибронектине, компонентах слизи (муцинах), покрывающих эпителиоциты, а также на других бактериях, прочно обосновавшихся в данной экологической нише. Адгезии препятствуют факторы, которые, экранируя бактериальные адгезины, мешают их взаимодействию с эпителиоцитами. Таким эффектом обладают секреторные IgA-антитела, структурные аналоги клеточных рецепторов, свободные муцины, фибронектин, лизоцим.
Адгезия — первый и ключевой этап стабилизации инфекта. За ней следует борьба за выживание и размножение бактерий. В стерильном кишечнике им требуется выстоять против попадающих туда секретов желудка, печени (желчи), поджелудочной железы, секреторных продуктов собственного эпителия. Бактериостатические и бактерицидные эффекты ослабевают по мере приближения к дистальным отделам тонкого кишечника, и, пройдя илеоцекальный угол (баугиниеву заслонку), бактерии попадают в комфортную зону. Появление микрофлоры меняет экологию кишечника, препятствуя или, напротив, способствуя размножению определенных групп бактерий. Сформировавшийся микробиоценоз существует как единое целое, как сообщество связанных цепями питания и микроэкологией индивидов, гомеостаз которого поддерживается внутренним взаимодействием его сочленов и жизнедеятельностью хозяина. Бактериальная адгезия отражается и на физиологии эпителиоцитов, индуцируя многие гены, от которых зависят нормальные взаимоотношения клеток с микрофлорой.
Слизистая оболочка толстого кишечника, главного резервуара бактерий, обильно инфильтрирована лимфоцитами, плазматическими клетками, макрофагами, т.е. по сути пребывает в состоянии хронического воспаления — неизбежного спутника нестерильных животных. Это такая же норма, как сама микрофлора, от которой избавлены лишь безмикробные животные, создаваемые искусственным путем. Воспалительный барьер препятствует реализации инвазивных потенций бактерий и, кроме того, служит источником биологически активных начал (в частности, цитокинов), которые включаются в регуляцию местного и общего гомеостаза. Снижение продукции цитокинов (интерферонов, факторов гемопоэза, хемокинов и пр.) — характерный признак безмикробных животных. С этим, в частности, могут быть связаны недоразвитие лимфоидной ткани, ослабление кроветворения и гипореактивность фагоцитов.
Физиологические функции нормальной микрофлоры кишечника
Тезис о жизненно важных функциях бактерий кишечника, провозглашенный Л. Пастером, абсолютно справедлив для травоядных, которые усваивают углеводы растительной пищи благодаря ферментативной (прежде всего целлюлазной) активности бактерий (рубец у жвачных животных, слепая кишка у лошади), но вовсе не однозначен для остальных животных, переваривающих пищу при помощи собственных ферментов пищеварительного тракта.
Безмикробные животные (серийные опыты над ними начались в конце 1940-х гг.) живут и размножаются не хуже обычных (конвенциональных). Их особенностями являются недоразвитие лимфоидной и гемопоэтической тканей, ослабление реактивности фагоцитов, гипогаммаглобулинемия, низкий уровень нормальных антител, истончение стенки кишечника. Это отражает снижение антигенной нагрузки и флогогенных стимулов, поддерживающих перманентное воспаление в слизистых оболочках нестерильных животных.
Нормальная микрофлора поддерживает иммунокомпетентные клетки в состоянии праймирования (субактивации), что обеспечивает более быстрый и эффективный ответ на инфекцию. Например, экспрессия HLA-2 (DR) на моноцитах новорожденного значительно ниже, чем у взрослого человека, а безмикробные животные по сходному признаку существенно уступают нестерильным особям. Это означает, что готовность клеток представлять антигены Т-хелперам усиливается под влиянием продуктов нормофлоры, что может быть связано с постоянной микробиндуцированной секрецией иммунотропных цитокинов. Недостатком продукции цитокинов (ростовых факторов) в субэпителиальной ткани кишечника можно объяснить и сниженный уровень гемо- и лимфопоэза безмикробных животных. Ослабление этого и других механизмов, незаметное в стерильных условиях, является серьезным минусом в обычной жизни, изобилующей столкновениями с потенциально опасными инфекционными агентами.
Одним из барьеров на пути экзогенных инфекций служит и сама микрофлора. Например, в чистой культуре шигеллы приживаются в кишечнике безмикробных мышей, но быстро элиминируются после добавления в корм кишечной палочки (конкурентная адгезия!?). Нормальная микрофлора участвует в обезвреживании токсинов, ограничивая болезнетворность токсигенных бактерий, попадающих в кишечник. С этим может быть связана более слабая активность ботулинического токсина при энтеральной инстилляции по сравнению с его парентеральным введением. Протективное значение имеют и так называемые нормальные антитела, образующиеся против антигенов нормальной микрофлоры. Некоторые из них перекрестно реагируют с патогенными бактериями, ослабляя их инвазивность. Полагают, например, что определенный вклад в возрастную эволюцию резистентности против палочки инфлюэнцы типа b, пневмококка и менингококка (они защищены от внутрисосудистого клиренса капсулой, обладающей низкой иммуногенностью для детей первых лет жизни) вносят антитела против антигенородственных полисахаридов кишечной палочки.
Бактерии кишечника (кишечная палочка, анаэробы) синтезируют витамины, в том числе биотин, рибофлавин, пантотеновую кислоту, пиридоксин, витамин К, фолиевую кислоту, витамины Е, В12. Однако почти каноническое представление о том, что бактерии обеспечивают снабжение хозяина витаминами, не бесспорно. Витамины не всасываются в толстом кишечнике, а потому можно рассчитывать только на те из них, которые в небольшом количестве образуются в подвздошной кишке. Это не очень надежный источник. Известно, что вегетарианцы, в рационе которых отсутствует витамин В12, чаще, чем обычные люди, страдают от его дефицита, хотя этот фактор и образуется в их кишечнике. Вряд ли имеют значение и другие потенциально полезные вещества, синтезируемые бактериями. Скорее всего, они выводятся наружу, не оказав ощутимого действия. Чтобы принести пользу, витамины и другие незаменимые метаболиты должны оказаться в тонком кишечнике, а это означает, что их главным источником служит пища. Некоторые животные (например, крысы) склонны к копрофагии (пожиранию собственных фекалий). В этом есть очевидный смысл: бактериальные метаболиты попадают в тонкий кишечник и утилизируются. Впрочем, косвенно мы тоже пользуемся витаминами и другими жизненно важными микробными продуктами. Так, единственным источником того же витамина В12 служат бактерии (главным образом анаэробы кишечника), и человек получает его с мясом животных, в тканях которых (особенно в печени) он накапливается.
Таким образом, мы приблизились к сформулированному выше убеждению И.И. Мечникова в том, что польза резидентных бактерий сводится прежде всего к защите от экзогенных инфекций. Проблема микробзависимого питания сомнительна и вряд ли имеет реальное значение.
Нормальная микрофлора: патология
Большинство бактерий, населяющих кишечник, относится к категории условно-патогенных, или микробов-оппортунистов, представляющих потенциальную опасность для хозяина. Парадоксально, но врачи чаще имеют дело с болезнями, причиной которых является нормальная флора, чем с патологией, вызванной внешней инфекцией. В отечественной медицине утвердилось понятие «дисбактериоз» (или «дисбиоз»), с которым связывают большинство негативных последствий, сопряженных с нарушениями микрофлоры, прежде всего кишечного микробиоценоза. Емкое по своей сути представление о дисбактериозах претендует на одну из базисных позиций в концепции о микробном симбиозе и его болезнетворных потенциях. Проблема обросла огромным числом клинических наблюдений и выглядит почти неприступной для критики. Впрочем, медицина знает немало примеров, когда разумные возражения тонули в эмпиризме практикующих врачей и его символической формуле — «видим эффект». Время разрушило немало подобных иллюзий, и первыми интерес к ним обычно теряли сами практики, только что восхвалявшие идею. Критика нужна для того, чтобы, избавившись от тенденциозности, утвердить рациональное начало, а затем усилить его. В таком анализе нуждается и концепция о дисбактериозе.
Факторы, влияющие на микрофлору кишечника
У здорового человека состав нормальной микрофлоры достаточно стабилен, если пренебречь такой «мелочью», как количественные колебания, которые для некоторых бактерий достигают нескольких порядков. Относительное постоянство отражает собственный (внутренний) гомеостаз нормальной микрофлоры, который базируется на динамической стабильности ее взаимоотношений с хозяином, т.е. с той средой, где микробы поддерживают свое существование. Эти взаимоотношения определяются постоянной эвакуацией содержимого кишечника (бактерии составляют около половины массы фекалий), быстрым обновлением эпителиального покрова слизистой оболочки (десквамация эпителия вместе с бактериями), физико-химическими параметрами, местными иммунными и воспалительными реакциями, которые сдерживают инвазию бактерий и (вместе с детоксицирующей функцией печени) нейтрализуют токсичные метаболиты бактерий. Кроме того, устойчивость микробиоценозов подвергается постоянным испытаниям со стороны внешней среды — непрерывного источника новых микробов, претендующих на закрепление в кишечнике.
Кишечная микрофлора довольно устойчива к переменам в диете. Это, очевидно, связано с тем, что независимо от исходного сырья в толстый кишечник попадает более или менее равноценный набор метаболитных отходов, необходимых для бактерий; они образуются при пищеварении в желудке и тонком кишечнике. Требуются специально спланированные диеты, чтобы дестабилизировать кишечный микробиоценоз. Пища, богатая углеводами, стимулирует бифидофлору и ведет к увеличению бактериальной массы толстого кишечника. Жировая диета угнетает бифидобактерии и энтерококки, но возбуждает размножение бактероидов. Белковая диета практически не влияет на спектр и количество кишечных бактерий. Эти и другие, по сути, экстремальные примеры лишь убеждают в относительной устойчивости нормофлоры. Пищевой дисбактериоз носит временный характер, исчезая при переходе на обычное питание. То же самое можно сказать о стрессорных дисбактериозах, регистрируемых при длительном пребывании в необычных условиях (космические полеты, зимовка в Антарктиде, работа в тяжелых условиях). Фактически это элемент адаптивных перестроек в организме. Такие дисбиотические реакции носят компенсаторный характер и легко устранимы. Микрофлора меняется у пожилых людей и зависит от времени года. Возрастной и сезонный дисбактериозы регистрируются у здоровых людей, что лишний раз говорит об условности нормы для кишечного микробиоценоза и чисто бактериологическом содержании понятия дисбактериоз.
Многочисленные наблюдения свидетельствуют об изменениях микробного пейзажа толстого кишечника при различных заболеваниях местного и общего характера. Дисбактериоз регистрируется у большинства больных с поражениями желудочно-кишечного тракта инфекционной и неинфекционной природы, у больных и реконвалесцентов после острых вирусных и бактериальных инфекций некишечной локализации, при хронических воспалительных и аллергических заболеваниях, лучевой болезни, у больных лейкозами и другими злокачественными процессами, при постлучевом синдроме, на фоне применения цитостатиков и антибиотиков. Лекарственные (особенно антибиотикозависимые) дисбактериозы отличаются наибольшей стабильностью и могут иметь серьезные последствия (классический пример — кандидомикоз).
Резюмируя, следует подчеркнуть, что сдвиги в нормальной микрофлоре всегда вторичны, отражают действие факторов, меняющих состояние кишечника или внутренний баланс в системе самого микробиоценоза.
Дисбактериоз как микробиологическое понятие
Формально определить дисбактериоз нетрудно: это любые, выходящие за рамки нормы (для данного биотопа) сдвиги в облигатной и/или факультативной микрофлоре. Увеличение количества факультативных микроорганизмов часто сочетается с расширением их видового спектра. К сожалению, приходится констатировать, что обследование фекалий на дисбактериоз нередко проводится без ясного понимания того, что дает подобный анализ и каким образом проецировать его на клинику, т.е. на профилактику и лечение заболеваний.
Анализ предусматривает обследование фекалий на количественное содержание бифидобактерий, лактобацилл, энтеробактерий (кишечной палочки и ее гемолитических вариантов, паракишечных (лактозодефектных) палочек, протея), энтерококка, золотистого стафилококка, синегнойной палочки, кандид. Акцент делается на снижении количества «благородных» бактерий (прежде всего бифидобактерий) и на повышении числа условно-патогенных видов. Трудности в трактовке результатов связаны с широкими колебаниями нормальных значений (т.е. тех же показателей у практически здоровых людей), быстрой сменяемостью нарушений, нестандартностью самого анализа. Немало примеров, когда дисбактериоз, установленный в одном учреждении, отвергается тестированием в других лабораториях. Не учитывается содержание бактероидов и других облигатных анаэробов, которые доминируют в нормальной микрофлоре кишечника, тем более что микрофлора фекалий — лишь приблизительная копия пристеночной, а тем более глубинной (криптовой) микрофлоры кишечника.
Тенденциозной выглядит трактовка понятия «условно-патогенные бактерии» в противопоставлении «сапрофитной флоре». Сапрофиты — экологическая категория, распространяющаяся на микроорганизмы, способные к автономному существованию в природе (свободноживущие формы). Альтернативой являются симбионты, не способные длительно пребывать во внешней среде (т.е. вне организма хозяина). И те и другие (сапрофиты и симбионты) могут обладать патогенностью (например, клостридии и синегнойная палочка — сапрофиты). Что касается условной патогенности, требующей для своего проявления местного или общего ослабления организма, то практически все представители нормальной микрофлоры обладают потенциальной болезнетворностью. Поэтому появление или количественное увеличение в фекалиях протея, синегнойной палочки, клебсиелл, энтеробактера, золотистого стафилококка и других бактерий не обязательно влечет за собой патологию. Неприятностей можно ожидать от обычной кишечной палочки, энтерококков, бактероидов, которые постоянно и в гораздо большем количестве содержатся в кишечнике. Опасность носит не видовой, а штаммовый (клональный) характер; в этом убеждают вспышки кишечных инфекций протейной, клебсиеллезной и другой природы. Не ясно и значение гемолитических штаммов кишечной палочки, появление которых считается одним из опасных признаков нарушения микрофлоры. Бактерии с гемолитической активностью широко представлены в нормальной микрофлоре; в кишечнике, например, их много среди бактероидов, спектр потенциально опасных факторов которых гораздо шире, чем у энтеробактерий. Энтеропатогенность эшерихий связана с их принадлежностью к определенным сероварам, что не учитывается в диагнозе на дисбактериоз.
В микробиологических описаниях принято говорить о степени кишечного дисбактериоза. Две первые из них отражают нарастание изменений в микрофлоре толстого кишечника (точнее фекалий), третья — отражает избыточное заселение бактериями вышележащих отделов пищеварительного тракта, четвертая — характеризует внекишечную контаминацию (проникновение микробов в кровь, мочевыводящую систему и т.д.). При этом предполагается, что дисбактериоз развивается как процесс, нарастая по мере воздействия факторов и условий, дестабилизирующих микрофлору. Однако такая последовательность необязательна. Избыточное размножение бактерий в тонком кишечнике и желудке не всегда сочетается с изменениями микробного пейзажа фекалий. Дидактично выглядит и концепция о генерализованных формах кишечного дисбактериоза. По сути, это видоизменение классической концепции об эндогенных, или аутоинфекциях, которые зарождаются в собственной микрофлоре при ослаблении местного и системного иммунитета. Причинная связь с нарушениями микробиоценоза кишечника вовсе не обязательна. Более того, источником этиологически сходных инфекций может быть окружающая среда, а потому говорить о внекишечных септических инфекциях, связанных с кишечной палочкой, протеем, бактероидами и другими бактериями, как о проявлениях кишечного дисбактериоза, представляется ненужным усложнением, которое лишь уводит от четкого понимания эпидемиологии, патогенеза и тактики лечения.
Перечисленные позиции говорят о противоречиях широко практикуемого ныне анализа на дисбактериоз кишечника. Фактически это дорогостоящее (тем более, что тестирование рекомендуется делать в динамике), трудоемкое исследование с невысокой (если не с нулевой) отдачей. Принимая во внимание огромный опыт такого рода исследований, представляется возможным почти безошибочно ставить регламентированный инструкциями диагноз «дисбактериоз», опираясь на клинику. Некоторые авторы пропагандируют дополнительные методы исследования — анаэробные техники, определение микробных метаболитов в кишечном содержимом и моче, газового состава выдыхаемого воздуха, некоторых персистентных характеристик условно-патогенной флоры и т.д. Это выглядит разумно, если не увлекаться декларациями. Иначе получится игра в методики все с тем же расплывчатым и неконструктивным диагнозом.
Дисбактериоз как патогенетическое понятие
Патогенетическое значение дисбактериозов обсуждается по трем основным направлениям:
1) патологический микробиоценоз как источник факторов, негативно влияющих на слизистую оболочку кишечника и вызывающих или поддерживающих развитие энтероколитов;
2) патологический микробиоценоз как источник факторов общетоксического действия;
3) патологический микробиоценоз как основа для развития эндогенных инфекций, выходящих за зону дисбактериоза. Проанализируем каждую из этих позиций. Одним из центральных симптомов энтероколитов является диарея. Она сопутствует многим функциональным и органическим поражениям кишечника. Острые энтериты и колиты наблюдаются при экзогенных инфекциях бактериальной, вирусной и протозойной природы. Концепция дисбактериоза претендует на расшифровку этиологически не ясных энтероколитных синдромов хронического или затяжного характера, которым сопутствуют более или менее выраженные изменения в облигатной и факультативной микрофлоре кишечника (фекалий). Это служит формальной основой для утверждения причинных связей между такого рода процессами и нетипичными для эубиоза энтеробактериями (гемолитическая кишечная палочка, протей, клебсиеллы и пр.), синегнойной палочкой, стафилококком, кандидами и др. Подобная идеология неоднократно заходила в тупик, но в таких случаях спасали беспроигрышные рассуждения о смешанной инфекции и многофакторном патогенетическом арсенале дисбиотической микрофлоры. Этиология ускользала, но патогенетическая канва и почва для спекуляций оставались.
Прорывом явилось утверждение представлений о клостридиальных поражениях толстого кишечника, клинический спектр которых варьирует от преходящей диареи до тяжелейшего псевдомембранозного колита. Их возбудителем является Clostridium difficile, факультативный представитель кишечной микрофлоры человека (см. «Клостридии»). Патология развивается на фоне применения антибиотиков и связана с ослаблением колонизационной резистентности кишечника на фоне дисбаланса в микробиоценозе. До признания этого факта этиологическая трактовка псевдомембранозного колита растворялась в концепции о дисбактериозе, которая, в принципе верно угадывая экологическую сущность процесса, не давала конкретных ориентиров в борьбе с этим заболеванием. Причины снижения колонизационной резистентности остаются неясными, но, по-видимому, связаны с извращениями анаэробной микрофлоры, ускользающими от рутинной диагностики. В частности, неожиданным и непонятным является антагонистический эффект дрожжей Saccharomyces boulardii. Это предложено использовать для лечения псевдомембранозного колита, и ряд фармакологических фирм уже отреагировали на эту информацию, наладив выпуск коммерческих препаратов на основе живой микробной массы S. boulardii. Повышенная восприимчивость к C. difficile не коррелирует с картиной классического дисбактериоза, а утвердившаяся клостридиальная инфекция не может быть ликвидирована при помощи общепринятых пробиотиков (эубиотиков).
Если толстый кишечник обычно страдает от дефицита резидентной микрофлоры и связанных с ней дефектов колонизационной резистентности, то для тонкого кишечника главной проблемой является избыточное размножение бактерий, оказывающих негативное воздействие на физиологию пищеварительного процесса. Это прежде всего связано с извращением метаболизма желчных кислот, необходимых для всасывания липидов и их производных. Желчные кислоты (холевая и хенодезоксихолевая) образуются в печени из холестерина и после конъюгации с глицином или таурином (производное цистеина) выделяются в 12-перстную кишку. Именно конъюгированные желчные кислоты играют решающую роль в абсорбции липидов эпителиальными клетками проксимальных отделов тонкого кишечника и их транспортировке в печень. Бактерии кишечника (бактероиды, энтерококки, лактобациллы, клостридии) вызывают деконъюгацию желчных кислот с образованием дериватов, которые лишены липотропной активности, не обеспечивая всасывания липидов. До 95% конъюгированных желчных кислот и их производных реабсорбируются в дистальных отделах тонкого кишечника и реутилизируются гепатоцитами в системе печеночно-кишечного круговорота. Лишь малая часть выделяется с фекалиями в виде бактериальных метаболитов, которые почти не всасываются в толстом кишечнике. Так как в норме проксимальные отделы тонкого кишечника бедны бактериями, то деконъюгации желчных кислот здесь практически не происходит. Это поддерживает нормальные условия для метаболизма липидов, которые успевают всосаться, не доходя до дистальных отделов подвздошной кишки, где имеется уже много бактерий, способных к инактивации желчных кислот.
Ситуация меняется при избыточном заселении бактериями тощей кишки. Опережающая деконъюгация желчных кислот, которая наблюдается в подобных случаях, ведет к нарушению всасывания липидов и развитию одного из патогенетических вариантов синдрома мальабсорбции. Возникает стеаторея (избыток непереваренных жиров в фекалиях) и диарея (результат торможения всасывания воды свободными желчными кислотами). Деконъюгированные желчные кислоты в большой концентрации поступают в воротную вену и, не успевая переработаться гепатоцитами, выходят в общую циркуляцию. Повышенная концентрация желчных кислот в кровотоке усугубляет нарушение обмена веществ. Формируется синдром контаминации тонкого кишечника (англ. bacterial overgrowth syndrom, т.е. синдром избыточного размножения бактерий) — единственное реальное проявление дисбактериоза тонкого кишечника. Из-за трудности забора материала для бактериологического анализа об этом судят по косвенным признакам, в частности по присутствию в моче бактериальных метаболитов (летучих фенолов). Антибиотики, снижая контаминацию тонкого кишечника, оказывают положительный эффект.
Согласно представлениям об отрицательном воздействии на хозяина бактериальных метаболитов толстого кишечника, допускалось, что, проходя через печеночный барьер, они вызывают хроническую интоксикацию, чреватую серьезными последствиями. Эта мысль была активно воспринята И.И. Мечниковым, который рассматривал толстый кишечник как средоточие зла, убеждая в необходимости эубиотической перестройки его микрофлоры. Особенно азартно он угрожал преждевременным старением, надеясь продлить жизнь через уничтожение или хотя бы ограничение гнилостной микрофлоры кишечника. Опыты на безмикробных животных подтверждают вероятность интестинальной интоксикации: стерильные крысы с искусственной непроходимостью кишечника живут в 5 раз дольше обычных (конвенциональных), а их перитонеальная жидкость не обладает летальным эффектом. Однако подобные опыты далеки от реальных ситуаций. Здоровый кишечник регулярно освобождается от бактерий и их метаболитов, что вместе с печеночным фильтром служит надежной защитой от токсичных продуктов микрофлоры. Никому не удалось доказать правильность идей И.И. Мечникова, а сама концепция аутоинтоксикации, исходящей из нормального кишечника, по сути не рассматривается.
Патология кишечника и/или печени меняет ситуацию, делая угрозу интестинальной интоксикации реальной. Кроме прямого воздействия токсичных метаболитов, следует помнить о возможности цитокинопосредованных эффектов микробных продуктов, прежде всего липополисахаридных эндотоксинов, которые в изобилии продуцируются грамотрицательными бактериями толстого кишечника. Физиологичные по своей природе, такие эффекты могут выходить за гомеостатические рамки, особенно при патологическом воспалении кишечника (мукозите), когда возникают условия для местной стимуляции большого числа клеток-мишеней. Ситуация становится еще напряженнее, если липополисахариды и функционально сходные молекулы выходят в кровь, получая доступ к резидентным (фиксированным) макрофагам печени, селезенки, легких. Это может спровоцировать синдром эндотоксинемии или, по крайней мере, его частные проявления.
Дестабилизационные процессы в кишечнике часто сочетаются с поражениями кожи, весьма произвольно классифицируемыми как аллергодерматозы, или атопический дерматит (доказать иммунологическую, т.е. истинно аллергическую природу заболевания удается не более, чем у трети больных). Этому обычно сопутствуют нарушения кишечной микрофлоры, что дает повод говорить о кожных проявлениях дисбактериоза. На вооружение нередко берется гистамин, продуцируемый бактериями кишечника. Это наивно, так как хозяин располагает огромными запасами собственного гистамина, а патогенез воспаления (в том числе аллергического) вовсе не исчерпывается этим медиатором. Кроме того, есть данные, что гистамин из пищеварительного тракта не всасывается в кровь, подвергаясь окислительному дезаминированию уже в полости кишечника. Наиболее общей причиной служит непереносимость пищевых продуктов, связанная с нарушением переваривания и всасывания. Комплексное лечение кишечника (оно благоприятно отражается и на его микрофлоре) дает положительные результаты.
Прослеживается известная преемственность между концепцией интестинальной интоксикации и гипотезой о связи между микрофлорой толстого кишечника и канцерогенезом. Она основана на наблюдениях о том, что бактерии толстого кишечника способны трансформировать эндогенные соединения и вещества, поступающие с пищей, в канцерогенные или коканцерогенные факторы, которые действуют на кишечник или, проникая в кровь, вызывают злокачественную трансформацию клеток других тканей. Примером является канцерогенная трансформация пищевых добавок типа цикломатов, стероидных гормонов, желчных кислот и др. Так, цикломат (циклогексамин сульфат) под влиянием бактериальных сульфатаз превращается в циклогексамин — сильный канцероген для эпителиоцитов мочевого пузыря. Значение подобных конверсий не известно, но они привлекают внимание современных онкологов.
Пожалуй, наиболее реальным является участие нормальной микрофлоры в развитии оппортунистических инфекций. Еще Ш. Николь говорил о «микробах выхода» (microbes de sortie), имея в виду условную патогенность большинства представителей эндогенной микрофлоры, которые способны активно или пассивно покидать место своей обычной дислокации, проникая в стерильные участки тела или внедряясь в чужие микробиоценозы. Это наблюдается в разных ситуациях: при перфорации стенки кишечника, повреждении кожи и слизистых оболочек, при восходящей инфекции мочевыводящей системы и пр. Вирулентность бактерий в сочетании с ослаблением местной и общей резистентности организма способствуют закреплению «микробов выхода» и реализации их патогенетического потенциала. Мы говорили об этом ранее с позиций общепринятой концепции оппортунистических инфекций. Некоторые из наиболее последовательных апологетов дисбактериоза относят к нему чуть ли не все оппортунистические инфекции.
При этом предполагается, что выходящие из-под контроля условно-патогенные бактерии гиперрепродуцируются не только в зоне своего естественного обитания, но и проникают в другие органы, инициируя инфекционный процесс. Вряд ли, однако, можно согласиться с тем, что, например, септическая инфекция мочевыводящих путей, вызванная кишечной палочкой, является результатом декомпенсированного дисбактериоза кишечника. Пиогенные бактерии, не дожидаясь дисбактериоза, постоянно проникают в систему мочевыделения из перианальной зоны, но быстро удаляются из мочевыводящего тракта, не успев спровоцировать воспалительную реакцию. Условия для их патогенетически значимого размножения возникают лишь при механической задержке мочи, травмировании слизистой оболочки диагностическими или лечебными процедурами, повышенном содержании в моче глюкозы (отличного продукта питания для бактерий). То же самое справедливо и для других «внекишечных проявлений дисбактериоза». Вряд ли, например, разумно лечить пиелонефрит или септическую инфекцию легких препаратами, нацеленными на восстановление микрофлоры кишечника. Если она и нарушена, то это вторичное явление, исчезающее вместе с ликвидацией базисного патологического процесса. Кстати, нечто подобное справедливо и для дисбактериоза самого кишечника. Некоторые авторы дифференцируют дисбактериозы кишечника по этиологическому принципу — протейный, клебсиеллезный, стафилококковый, бактероидный, синегнойный и пр. Но если этиологическая значимость соответствующих бактерий в кишечных расстройствах больного доказана (что сделать довольно трудно), проще говорить об инфекции, а не о дисбактериозе и лечить пациента, исходя из общих принципов антибактериальной терапии.
Формально любую экзогенную инфекцию можно рассматривать как дисбактериоз или дисбиоз, так как всякое экзогенное заражение сопровождается появлением в организме количественно и/или качественно новых микробиоценозов, т.е. изменением микрофлоры. Безусловно, дисбактериоз как один из механизмов нарушения колонизационной резистентности может стать основой для оппортунистических инфекций кишечника (как это установлено для C. difficile), но внекишечная локализация «кишечных бактерий» не обязательно является фазой местного дисбиотического процесса. Скорее наоборот: перекосы в микрофлоре, регистрируемые в таких случаях, возникают как вторичная реакция на изменения экологии кишечника, спровоцированные дистантным инфекционным процессом. Иными словами, являясь реальной микробиологической категорией, кишечный дисбактериоз несет в подобных ситуациях сомнительную патогенетическую нагрузку.
Дисбактериоз как индикатор здоровья
В наиболее общем виде можно говорить о двух механизмах нарушения нормальной микрофлоры. Один из них связан с выбиванием бактерий антимикробными препаратами, прежде всего антибиотиками. Второй является следствием реактивной перестройки микрофлоры в ответ на изменения той среды, где сосредоточен данный микробиоценоз. Как уже отмечалось, сдвиги в микрофлоре кишечника наблюдаются при многих заболеваниях, в том числе внекишечной локализации. Это означает, что кишечник меняет свою экологию, реагируя на разнообразные нарушения гомеостаза. Отсюда дисбиотические реакции следует считать косвенным отражением такого рода дестабилизационных процессов. Они не ограничиваются перекосами в микрофлоре кишечника, но проявляются и в других участках тела. Это объясняется тем, что изменение состояния организма в той или иной мере сказывается на всех отделах слизистой оболочки и кожи, обеспечивая синхронность дисбиотических реакций разной локализации, а также пассивных сдвигов в микрофлоре, возникающих на фоне антибактериальной терапии. Парадокс, но заключение о норме или патологии микробиоценоза толстого кишечника с известной долей вероятности можно сделать по результатам анализа микрофлоры ротоглотки, буккального эпителия и даже кожи.
В целом, перестройку в нормальной микрофлоре можно использовать как критерий для суждения о состоянии здоровья, его явных и скрытых нарушениях, о позитивных и негативных тенденциях в развитии патологического процесса и адекватности проводимой терапии. Громоздкость бактериологического анализа фекальной микрофлоры практически исключает такую возможность, тем более что наблюдение необходимо вести в динамике. В связи с этим представляет интерес тестирование микробиоценозов других участков тела. В свое время широко практиковалось изучение микрофлоры кожи при помощи удобного технического приема, предложенного Н.Н. Клемпарской. Нами используется тест на естественную колонизацию буккального эпителия, который в норме обильно заселен оральными стрептококками. Снижение этого показателя и обсеменение буккальных эпителиоцитов посторонней микрофлорой наблюдается при различных заболеваниях, сигнализируя о дестабилизации гомеостаза на уровне слизистых оболочек (см. рисунок).

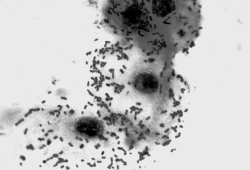
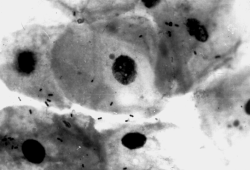
Естественная колонизация буккального эпителия: а — здоровый донор (12 лет), эпителиоциты обильно колонизированы оральными стрептококками; б — ребенок (4 года) с бронхиальной астмой, резкое снижение количества оральных стрептококков. х630 (препараты М.А. Абаджиди)
Дисбактериоз как объект терапии
Один из принципов утилитарной врачебной логики состоит в том, что все показатели, отличающиеся от условной нормы, требуют коррекции. При этом не учитывается вторичность многих сдвигов, имеющих скорее адаптивный, чем патогенетический характер. Ликвидация базисного процесса автоматически устраняет такого рода псевдонарушения. По сути это символы патологии, а не патогенетические детерминанты, требующие врачевания. Именно с таких позиций следует подходить к целесообразности направленной коррекции дисбиотических состояний, которые сопутствуют многим нарушениям гомеостаза.
Необходимы критерии, по которым можно было бы судить о том, когда дисбиозы переходят грань адаптивной реакции и трансформируются в патогенетически значимый механизм. Собственно на это нацелены все классификации кишечного дисбактериоза, подчеркивающие опасность глубоких перестроек резидентной флоры. Наиболее логичной выглядит заместительная терапия живыми бактериями, населяющими толстый кишечник. Препараты, изготовленные на их основе, получили название пробиотиков, или эубиотиков, так как они нацелены на восстановление эубиотического (нормального) состояния микрофлоры. В коммерческом варианте такие препараты доступны в виде колибактерина (живая кишечная палочка), бифидумбактерина (бифидобактерии), лактобактерина (лактобациллы) и их комбинаций (бификол, бифилакт); в последнее время стал рекламироваться препарат на основе энтерококка. Все они применяются в форме лиофилизированных живых бактерий либо в виде продуктов, приготовленных путем сбраживания молока соответствующими бактериями. Много усилий потрачено на поиски оптимальных штаммов, обладающих антагонистическими (по отношению к потенциально опасным бактериям) свойствами, устойчивостью к антибиотикам, активно сбраживающих молоко и пр. Общая идея сводится к искусственному заселению кишечника «благородными» бактериями, вытеснению благодаря этому болезнетворных штаммов и восстановлению нормального микробиоценоза (эубиоза). Огромный опыт, зафиксированный в необозримом количестве публикаций, свидетельствует о положительном эффекте бактериотерапии при хронических заболеваниях кишечника и их системных осложнениях (например, при аллергодерматозах). С такой же целью и с неменьшим успехом предложено использовать добавки к питанию (пробиотики), стимулирующие размножение полезных бактерий, прежде всего бифидобактерий (бифидогенные факторы или бифидогенный режим).
Несмотря на очевидную логику, механизм действия пробиотиков остается неясным. Прежде всего не известно, насколько активно они ведут себя в кишечнике, а следовательно, в какой степени проявляют свои полезные свойства, по которым их отбирают in vitro. По крайней мере, после прекращения поддерживающей терапии искусственно введенные штаммы быстро исчезают из кишечника и замещаются случайной микрофлорой. Вызывает удивление, что хвалебным отзывам об эффективности препаратов, приготовленных на основе живых бифидобактерий, предшествовал не меньший восторг от применения кишечной палочки (мутафлор A. Nissle и колибактерин Л.Г. Перетца), хотя по количественному содержанию в нормальной микрофлоре они значительно уступают анаэробам. То же самое можно сказать о лактобактерине, который высоко ценился еще И.И. Мечниковым и его последователями. Кстати, выбор пробиотика на основании данных бактериологического анализа (например, назначение бифидумбактерина при дефиците бифидобактерий, а лактобактерина при недостатке лактобацилл) — не более чем иллюзия. Опыт показывает, что клиническая эффективность не коррелирует с подобной логикой. То же самое справедливо для пробиотика Saccharomyces boulardii и препаратов, в состав которых входят аэробные спорообразующие бактерии, не являющиеся представителями нормальной микрофлоры кишечника — бактисубтил (Bacillus spp.) и споробактерин (B. subtilis). Попытки объяснить их положительное влияние на кишечный эубиоз только начинаются и уж никак не связаны с заместительным эффектом.
Благотворное влияние продуктов молочнокислого брожения при заболеваниях кишечника известно с незапамятных времен, и с этой точки зрения эффект молочных продуктов, заквашенных бифидобактериями и лактобациллами, не является неожиданностью. Вопрос лишь в том, в какой степени это бактериотерапия (т.е. эффект живых бактерий, искусственно трансплантируемых в кишечник), а в какой — диетотерапия (т.е. действие комплекса преформированных полезных факторов). Кстати, еще до И.И. Мечникова увлекались молочной кислотой все с тем же «великолепным результатом». Истина, по-видимому, в том, что лечить надо прежде кишечник, а уже потом его микрофлору, исходя из принципа, что дисбактериоз — болезнь организма, а не микрофлоры, и как вторичное явление спонтанно обратим. Впрочем, так фактически и поступают: бактериотерапия входит в комплекс лечебных мероприятий, и отыскать первичные и последующие звенья в этой комплексности столь же непросто, как и в механизмах (символах?!) самого патологического процесса. Патология кишечника предрасполагает к нарушениям микрофлоры, а они в свою очередь могут негативно влиять на функции кишечника, от которых зависит норма микробиоценоза.
Патогенетическое лидерство в реализации этого порочного круга принадлежит оппортунистическим инфекциям, которые возникают при активации эндогенных бактерий или развиваются при экзогенном инфицировании. Примерами являются клостридиальный (C. difficile) колит и кандидомикоз. Значение других «дисбактериозных» бактерий (золотистый стафилококк, атипичные эшерихии, протей и другие энтеробактерии, синегнойная палочка) менее очевидно, хотя часто и воспринимается как аксиома. В подобных случаях в лечебные схемы вводятся антимикробные химиопрепараты, и терапия становится, по сути, этиотропной, т.е. дисбактериоз лечат как инфекционное заболевание. Этому трудно отказать в логике за единственным исключением: специфичность антимикробных средств относительна и попутно неизбежно страдает резидентная микрофлора. Такая тактика не отвечает принципам лечения дисбиотических состояний и должна быть ограничена жизненными показаниями (например, тяжелые формы клостридиального колита) или, по крайней мере, ситуациями, где патогенетическая значимость инфекции достаточно обоснована (например, синдром контаминации тонкого кишечника). Дисбактериоз — понятие микробиологическое, и требуется серьезно подумать, прежде чем проецировать его на клинику. Это, в частности, относится к диагнозам типа «протейный», «стафилококковый», «клебсиеллезный» и другие дисбактериозы. Они имеют сомнительное значение и не должны вызывать чрезмерных опасений, настраивая врачей на немедленную этиотропную терапию (не путать с острыми кишечными инфекциями, где роль энтеропатогенных штаммов тех же видов бактерий вполне доказуема).
Следует сказать об еще одной иллюзии, которая внешне выглядит как блестящая антибактериальная стратегия. Речь идет о фаготерапии, нацеленной на уничтожение в кишечнике протея, клебсиелл, синегнойной палочки, стафилококка, атипичных эшерихий. Обладая селективным (клональным) бактерицидным эффектом, бактериофаги идеально отвечают критерию П. Эрлиха о «волшебной пуле». Казалось, открытие фагов сулит блестящую перспективу для борьбы с инфекционными заболеваниями, тем более что (как нередко бывает) первые результаты выглядели блестяще. Но вскоре пришло разочарование: фаготерапия и фагопрофилактика не оправдали возлагавшихся на них надежд. В этом есть биологические причины (слишком узкая специфичность фагов, быстрое появление фагорезистентных штаммов), которые в принципе можно преодолеть, располагая достаточно большой коллекцией фагов и адаптируя их к выделяемым культурам. Но есть и возражения технического характера. Например, фаги плохо сорбируются бактериями в коллоидной среде, так что трудно представить возможность реализации их бактерицидности в толстом кишечнике, где бактерии окутаны слизью и перемешаны с фекальными массами. Проблемой является доставка фагов к месту назначения. В частности, практикуемое введение препаратов при помощи клизмы не обеспечивает поступления фагов в проксимальные отделы толстого кишечника, где может быть локализован основной дисбиотический процесс. Еще наивнее выглядит введение фагов в виде ректальных свечей. Словом, даже блестящая активность фага in vitro не гарантирует его эффективности при введении в организм.
В этой связи отметим удивительные (лучше сказать — странные) работы, где сообщалось об эффективности парентерального введения фагов для лечения очаговых пиогенных процессов (их было немало в прошлом). Не понятно, как авторы представляли себе маршрут фагов до их потенциальной мишени после такого рода инстилляций. Совершенно очевидно, что они должны были элиминироваться механизмами, обеспечивающими клиренс тканевой жидкости и крови от чужеродного материала, тем более, что при повторных инъекциях фаги, являясь отличными антигенами, нейтрализуются антителами.
Забывалось (и забывается) то, что коммерческие фаги представляют собой взвесь фаговых частиц в белково-пептонных средах с примесью значительного количества продуктов (в том числе биологически активных), образующихся при фагозависимом лизисе бактерий. Более того, фаги составляют лишь небольшую часть этого сложного комплекса. Отсюда введение такого рода препаратов следует рассматривать не только как инстилляцию самого фага, но прежде всего как введение чужеродного белка и бактериальных адъювантов, т.е. по сути как воскрешение почти забытой (но некогда популярной) протеинотерапии и повторение тактики биостимуляции, основанной на использовании производных бактерий типа липополисахаридов (пирогенал, продигиозан), пептидогликанов и пр. Именно неспецифическим потенцированием резистентности организма можно объяснить удачи парентеральной фаготерапии хронических пиогенных инфекций (кстати, многие авторы отмечали осложнения, характерные для введения чужеродного белка и бактериальных биостимуляторов — лихорадку, воспалительные отеки и пр.). Фаги предложено использовать для орошения ран с целью профилактики и лечения септических инфекций. В этом случае также не учитывается возможность неспецифического эффекта, а именно воспалительной реакции в ответ на инстилляцию чужеродного белка.
Не исключено, что элементы неспецифического флогогенного эффекта имеют место и при пероральной бактериотерапии. В данном случае одномоментно вводятся огромные дозировки бактерий, с которыми организм никогда не сталкивается в естественных условиях. Если учесть перманентное воспаление, в котором находится слизистая оболочка тонкого кишечника, можно представить, что столкновение с искусственно введенными бактериальными продуктами вызовет реакцию эффекторов воспаления и иммунитета с высвобождением медиаторов широкого спектра местного и общего действия.
Установлено нормализующее влияние различных пробиотиков на функциональный статус лимфоцитов, образование иммуноглобулинов, цитокинов, транскрипционных факторов эпителиальных клеток, состояние эпителиального барьера и пр. Все это способствует восстановлению физиологических взаимоотношений с нормальной микрофлорой, в частности ведет к повышению колонизационной резистентности слизистых оболочек против типовых проявлений инфекционного процесса. Примером является воспаление, обретающее характер порочного круга, который поддерживается за счет функциональных нарушений эпителиоцитов. Принципиальная задача — разорвать порочный круг, сделав процесс обратимым. Именно на это направлено применение пробиотиков, которое сопровождается нормализацией микроэкологии кишечника и факторов местного иммунитета.
Представление о живых пробиотиках как о факторах, колонизирующих кишечник и тем самым излечивающих дисбактериоз, в лучшем случае наивно. Об этом говорит хотя бы то, что позитивные (профилактические и лечебные) эффекты могут быть воспроизведены с убитыми бактериями и их дериватами. Это допускает более широкое толкование термина, относя к пробиотикам не только живые, но также инактивированные бактерии и их субкомпоненты, позитивно влияющие на здоровье.
Сепсис
Сепсис — проблема не микробиологическая,
а макробиологическая. И.В. Давыдовский
- Септический синдром и воспаление.
- Бактериемия и интоксикация.
- Сепсис и реактивность.
- Пусковые механизмы патогенеза.
- Флогогенные цитокины.
- Терапия: иллюзии и реальность.
В переводе с греческого сепсис означает гниение. Так когда-то называли любую гнойную инфекцию — сепсис кожи, сепсис слизистых оболочек, септические раны и пр. Словосочетание «септические инфекции» и сегодня фигурирует в практической медицине, хотя правильнее говорить о пиогенных инфекциях, сохранив за сепсисом понятие об особом синдроме, который возникает при генерализованной воспалительной реакции в ответ на внедрение в кровь живых возбудителей (бактериемия) или их продуктов (токсинемия). Последнее означает, что заражение крови необязательно для развития септического синдрома. Это реально отражает микробиологическую сущность процесса, который может протекать в двух вариантах — сепсис с заражением и без заражения крови.
Представления о генерализованном (внутрисосудистом) воспалении позволили объединить на единой патогенетической основе группу патологий, у которых формально мало общего. Кроме инфекционных заболеваний к ним относятся тяжелая травма, острый панкреатит, осложнения после гемодиализа, экстракорпорального кровообращения и других вмешательств в систему внутрисосудистого гомеостаза. Хотя септический синдром может осложнять любые инфекции, он чаще связан с пиогенными бактериями. В основном это госпитальная патология. Проблема условной патогенности и механизмов ее реализации не решена до сих пор. Здесь ярко сфокусирована диалектика оппортунистических инфекций и клинической микробиологии в целом: причиной тяжелейших осложнений могут быть микробы, безвредные для здоровых людей. Среди них доминируют облигатные или факультативные представители нормальной микрофлоры — кишечная палочка, стафилококки, псевдомонады, бактероиды, энтерококки; единичные случаи могут быть связаны с любыми (особенно грамотрицательными) бактериями.
Есть злая ирония в том, что успехи медицины (в частности, широкое применение инвазивных манипуляций) повысили вероятность госпитального сепсиса, обеспечив ему одну из лидирующих позиций среди причин внутрибольничной смертности. Это связано с расширением групп риска за счет факторов, отрицательно влияющих на местную и общую резистентность организма. Основные из них можно суммировать следующим образом:
1) повреждение кожи и слизистых оболочек (раны, ожоги, пролежни, катетеризация и инструментальное обследование мочевыводящих путей, бронхоскопия, внутрисосудистые катетеры и пр.);
2) гипотрофия стенки кишечника, в частности от применения цитостатиков (подавление пролиферации быстро обновляющихся эпителиоцитов слизистой оболочки);
3) гранулоцитопения на фоне применения цитостатиков (торможение пролиферации костномозговых предшественников);
4) гипореактивность клеток-эффекторов воспаления (новорожденные, больные диабетом, циррозом печени);
5) гипогаммаглобулинемия, врожденные дефекты фагоцитов и комплемента;
6) дефекты лимфоцитов, индуцированные иммунодепрессантами и цитостатиками;
7) спленэктомия или плохо функционирующая селезенка (например, больные с серповидноклеточной анемией), при которых характерна склонность к молниеносному сепсису от инкапсулированных бактерий (пневмококк, палочка инфлюэнцы, менингококк). Транзиторная бактериемия — достаточно заурядное явление. Бактерии нередко попадают в кровь из нормальной микрофлоры после процедур, нарушающих гематотканевые барьеры (табл. 1). Кровь способна к самоочищению, и обычно микробные дериваты быстро удаляются из циркуляции (см. «Механизмы противоинфекционного иммунитета»). К септическим осложнениям предрасполагают высокая вирулентность возбудителя, массивная доза инфекта, его восполнение за счет местных очагов инфекции, закрепление бактерий на поврежденных участках эндотелия, внутрисосудистых протезах, тромбах, дефекты внутрисосудистого клиренса.
Таблица 1
Вероятность транзиторной бактериемии после различных процедур
| Процедура/манипуляция | Число позитивных гемокультур*, КЕ |
| Удаление зуба | 18—85 |
| Жевание конфет или парафина | 17—51 |
| Чистка зубов | 0—26 |
| Периодонтальные операции | 32—88 |
| Бронхоскопия | 15 |
| Тонзиллэктомия | 28—38 |
| Назотрахеальная интубация | 16 |
| Гастроинтестинальная эндоскопия | 8—12 |
| Сигмоидальная эндоскопия | 0—9,5 |
| Бариевая клизма | 11 |
| Перкутанная биопсия печени | 3—13 |
| Бужирование уретры | 18—33 |
| Катетеризация уретры | 8 |
| Цистоскопия | 0—17 |
| Трансуретральная резекция простаты | 12—46 |
| Нормальные роды | 0—11 |
| Биопсия шейки матки | 0 |
| Удаление/введение внутриматочных спиралей | 0 |
*Уровень бактериемии обычно не превышает 10 колониеобразующих единиц в 1 мл. Вероятность возрастает при повышенной обсемененности слизистых оболочек, например при воспалительных заболеваниях.
Патогенетически значимая бактериемия может эволюционировать по-разному:
- 1) бактерии разносятся по организму, инициируя новые очаги воспаления (септикопиемия);
- 2) возбудитель выделяет септикогенные факторы после закрепления внутри сосудистого русла (участки поврежденного эндотелия, тромбы, внутрисосудистые протезы);
- 3) бактерии размножаются в кровяном русле (септицемия), создавая опасность молниеносных осложнений.
Последний вариант (точнее, его микробиологическая трактовка) признается не всеми авторами. Есть предложение вообще отказаться от понятия септицемия, тем более что в переводе это означает «гноекровие», т.е. по сути «сепсис крови». Чтобы избежать противоречий, понятие «септицемия» можно оставить для обозначения септических осложнений, развивающихся при заражении крови, а категорию «сепсис» использовать как более широкое микробиологическое понятие, для которого бактериемия не обязательна.
Каким бы способом ни развивалась инфекция, ее трансформация в сепсис зависит от реактивности организма. Суть в том, что эффекторы воспаления, не всегда способные остановить процесс на местном уровне, активно реагируют на флогогенные стимулы, превращаясь в инструмент системной патологии. С этой точки зрения сепсис и очаговая инфекция — качественно сходные категории, которые базируются на однотипных механизмах (воспалительная реакция) и различаются лишь по степени развития. Это соответствует классической формуле, согласно которой сепсис есть продолжение местного пиогенного процесса. Действительно, очаговая инфекция обычно предшествует и/или сопутствует септическим осложнениям. Но нередко эта связь теряется, и, отрываясь от пускового механизма, сепсис начинает жить собственной жизнью. В таких случаях удаление пиогенного очага не спасает больного. Это, а также то, что клиника сепсиса мало зависит от специфики возбудителя, постоянно смущало патологов. И.В. Давыдовский считал сепсис макробиологической, а не микробиологической проблемой. Другой крупный отечественный патолог А.И. Абрикосов говорил, что сепсис — не продукт особых свойств инфекции, а результат особого состояния организма. Вспомним и Л. Пастера, который, не побоявшись унизить свое детище (микробную теорию медицины), заявил на склоне лет: «Микроб ничто, субстрат (т.е. организм) все!».
Признание важности реактивных сдвигов для развития септического процесса долго оставалось декларацией. Лишь в последнее время в связи с детализацией знаний о молекулярных основах воспаления эта позиция стала обретать конкретные очертания. Суммируя классические и современные взгляды, сепсис (септический синдром) можно рассматривать как неспецифическую микробную интоксикацию, опосредованную через дестабилизацию потенциальных эффекторов воспаления, подобно тому как анафилактические реакции возникают при антигениндуцированной активации тучных клеток. Именно гиперпродукция эндогенных медиаторов, побуждаемая микробными дериватами, служит причиной разнообразных расстройств, которые могут привести к тяжелой полиорганной недостаточности (сепсис не щадит ни одного органа!) и фатальному извращению гемодинамики (септический шок) (табл. 2).
Таблица 2
Возможные медиаторы сепсиса
| Фактор | Патофизиологические эффекты (точка приложения) |
| Цитокины (туморонекротический фактор-альфа, интерлейкины-1, -6, -8) | Имитация септического синдрома и его частных признаков |
| Фактор Хагемана (фактор XII), тканевой фактор (внешний тромбопластин), фактор Х | Гемокоагуляция, фибринолиз |
| Система комплемента | Хемотаксис, адгезия и агрегация нейтрофилов, утечка из капилляров |
| Эндорфины | Гипотензия |
| Лейкотриены и тромбоксаны | Агрегация тромбоцитов, адгезия нейтрофилов, утечка из капилляров, ослабление сократимости миокарда |
| Простагландины (особенно Е2 и I2) | Гипотензия, адгезия нейтрофилов на эндотелии, лихорадка, мышечные боли, протеолиз мышц |
| Брадикинин (через фактор XII и калликреин) | Гипотензия, утечка из капилляров |
| Серотонин | Легочная гипертензия, утечка из капилляров |
| Гистамин | Гипотензия, утечка из капилляров |
| Фактор активации тромбоцитов | Гипотензия, утечка из капилляров, агрегация тромбоцитов, активация лейкоцитов, ослабление сократимости миокарда |
| Продукты фагоцитов — гидролазы, катионные белки, кислородные радикалы | Повреждение эндотелия, утечка из капилляров |
| Миокардиальный депрессант | Ослабление сократимости миокарда |
| Эндотелин-1 | Вазоконстрикция (особенно в почках) |
| Оксид азота (фактор релаксации эндотелия) | Гипотензия |
Развитие септического процесса базируется на двух главных патофизиологических механизмах — повышении проницаемости микрососудов и склонности к внутрисосудистой гемокоагуляции. Центральным звеном, от которого зависят все последующие события, является поражение эндотелия. Под этим следует понимать не только структурное повреждение (признак далеко зашедшего процесса), но и несбалансированную активацию эндотелиоцитов, выводящую их из-под гомеостатического контроля. На эндотелиальных клетках возрастает число рецепторов для адгезивных молекул лейкоцитов. Это прежде всего сказывается на взаимоотношениях с нейтрофилами. Подвергаясь активации, они повреждают сосудистую стенку, увеличивая ее проницаемость для плазмы. Утечка из капилляров создает основу для падения кровяного давления, нарушения кровоснабжения и кислородного голодания (в финальной стадии — катастрофического) тканей.
Особенно драматичны события в легких, так как именно здесь секвестрируется большинство нейтрофилов. Острый респираторный дистресс-синдром доминирует в спектре многочисленных органных поражений при сепсисе. Он выделен в самостоятельную нозологическую единицу в 1960-х годах, объединив разрозненные наблюдения о дыхательной недостаточности некардиогенной природы — шоковое легкое, травматический отек легких, некардиогенный отек легких, посттрансфузионное легкое, массивный коллапс легких. В основе синдрома лежит повышенная проницаемость альвеолярных капилляров, обусловленная диффузным повреждением активированными нейтрофилами микроциркуляторного ложа. Деструкция, захватывающая стенку альвеол, вызывает накопление внутриальвеолярного экссудата, сокращая дыхательную поверхность легких.
Гиперактивация эндотелиоцитов отражается и на гомеостазе плазмы, повышая склонность к тромбообразованию. При ослаблении контролирующих рычагов это ведет к одному из ключевых проявлений сепсиса — диссеминированной коагулопатии.
Усиление тромбогенной функции эндотелия связано по меньшей мере с тремя причинами.
Во-первых, подобно другим клеткам, эндотелиоциты содержат прокоагулянт (внешний тромбопластин, или тканевой фактор), образование которого многократно усиливается при активации. В этом отношении с ними могут конкурировать лишь макрофаги и нейтрофилы, которые тоже способны к реактивному выбросу тромбогенных начал.
Во-вторых, на поверхности эндотелиальных клеток имеется тромбомодулин — белок, обладающий свойствами антикоагулянта. В отличие от тромбопластина его синтез снижается при стимуляции, в том числе флогогенными цитокинами.
Наконец, эндотелиоциты продуцируют фактор, блокирующий активацию плазминогена, секреция которого, как и тромбопластина, возрастает при активации эндотелиальных клеток.
В целом стимулированные эндотелиоциты являются зоной риска по тромбообразованию, угрожая местными осложнениями и системной гемокоагуляцией. Этому способствует и усиление рецепторзависимой адгезии тромбоцитов с подключением внутреннего (тромбоцитарного) механизма свертывания крови. Возможен порочный круг: тромбин, образующийся при участии стимулированного эндотелия, активирует эндотелиоциты, стабилизируя синтез собственного индуктора — эндотелиоцитарного тромбопластина.
Классическим индуктором септического синдрома является липополисахаридный эндотоксин (ЛПС) грамотрицательных бактерий (у грамположительных бактерий эту роль, хотя и менее успешно, выполняют пептидогликан и тейхоевые кислоты). Попадая в кровь, ЛПС втягивает в реакцию множество мишеней, активируя лейкоциты, тромбоциты, эндотелиоциты, комплемент и др. Не случайно патогенетические схемы ЛПС-интоксикации перегружены деталями, иллюстрирующими изобилие эффектов ЛПС в системах воспаления и иммунитета. Высказывалось предположение, что все начинается с комплемента (активация по альтернативному пути), производные которого (прежде всего С5а) растормаживают последующие звенья септического каскада. Современная позиция менее прямолинейна: главным источником пусковых факторов считается не плазма, а активированные клетки.
Идея о внутрисосудистом воспалении стимулировала развитие представлений о физиологических токсинах, которые опосредуют дестабилизирующий эффект экзогенных флогогенов в каскаде межклеточных и гуморально-клеточных взаимодействий. Наиболее логично это отражено в концепции о флогогенных цитокинах (см. «Механизмы противоинфекционного иммунитета», «Воспаление и иммунитет»). Обобщенная формула гласит, что в основе септической патологии лежит неконтролируемый дисбаланс («метаболическая анархия») в системе цитокинов и их ингибиторов.
Как уже говорилось, способностью к продукции цитокинов наделены многие клетки, но далеко не все из них причастны к инициации септического синдрома. Для этого они должны отвечать по меньшей мере двум условиям:
- продуцировать патогенетически значимое количество флогогенных (септикогенных) цитокинов;
- оперативно секретировать их в кровяное русло.
Первыми в реакцию на внутрисосудистое внедрение чужеродных агентов включаются макрофаги, фиксированные в строме печени, легких, селезенки, костного мозга. При стимуляции они секретируют множество биологически активных молекул, в том числе большинство из известных цитокинов. Это позволяет думать, что именно активированные макрофаги служат опережающим источником септикогенных молекул. Наряду с этим имеют значение и макрофагальные цитокины (монокины), которые поступают в кровь непосредственно из очагов воспаления.
Секреция цитокинов неодинаково выражена при клиренсе различных материалов. Это связано с особенностями активации макрофагов через разные рецепторы, влияя на развитие септического синдрома. К примеру, ЛПС воспринимаются несколькими клеточными рецепторами, но лишь подключение CD14 и тольподобных рецепторов (TLR) типа 4 обеспечивает выраженный секреторный ответ макрофагов на малые концентрации ЛПС. В этой реакции участвуют особые острофазные белки сыворотки, которые, соединяясь с липидным фрагментом ЛПС, опсонизируют его для взаимодействия с клетками. ЛПС-связывающие белки инициируют секреторный ответ макрофагов при клинически значимой эндотоксинемии. Это обеспечивает септикогенность минимальных дозировок ЛПС и лидирующую роль макрофагов в патогенезе ЛПС-индуцированных осложнений.
Из обширного арсенала цитокинов в нашем контексте наибольший интерес представляют туморонекротический фактор-альфа (ТНФ-a), интерлейкины-1, -6, -8 и гамма-интерферон. Приоритет отдается ТНФ-a. Его главный источник — макрофаги, но ТНФ-a-рецепторы есть на всех клетках за исключением эритроцитов. Это определяет необыкновенно широкий спектр физиологических и патофизиологических эффектов ТНФ-a (табл. 3). Внутривенное введение ТНФ-a воспроизводит многие признаки сепсиса, включая нейтропению, отек легких, падение кровяного давления. Сходным действием обладает интерлейкин-1, но по кинетике накопления в крови после введения ЛПС он отстает от ТНФ-a. Важно и то, что действие ТНФ-a на чувствительные клетки продолжается в течение 24—36 ч. Это означает, что реакция фактически трансформируется в процесс, когда мишень поддерживается в активированном состоянии после элиминации пускового агента. В эксперименте антитела против ТНФ-a блокируют септические симптомы и защищают от летальных исходов грамотрицательной бактериемии.
Таблица 3
Биологическая активность цитокинов, связанных с развитием септического синдрома
| Цитокин | Основные эффекты |
| ТНФ-a | Повышение адгезивности (усиление экспрессии адгезивных молекул) эндотелиальных клеток, нейтрофилов, эозинофилов, базофилов, моноцитов и (в меньшей степени) лимфоцитов Повышение тромбогенной (усиление продукции внешнего тромбопластина) и ослабление антикоагулянтной (ослабление экспрессии поверхностного тромбомодулина) активности эндотелиальных клеток Повышение проницаемости сосудов (прямая токсичность для эндотелиальных клеток) Стимуляция синтеза острофазных белков (активация гепатоцитов) Пирогенность (прямое действие на гипоталамус) Активация макрофагов (усиление продукции интерлейкинов-1, -6, -8, фактора активации тромбоцитов, лейкотриенов, тромбоксана А2, простагландинов) Усиление мобилизации костномозговых предшественников нейтрофилов; повышение фагоцитарной активности нейтрофилов Усиление секреции коллагеназы; вариабельные эффекты на пролиферацию фибробластов (в зависимости от присутствия других медиаторов) Снижение трансмембранного потенциала мышечных клеток и ослабление сократительной активности кардиомиоцитов Снижение кровяного давления, шок Индукция молекул главного комплекса гистосовместимости класса 1 (HLA-1)Подавление активности липопротеинлипазы и метаболизма жировых клеток; истощение энергетического потенциала мышечных клетокСтимуляция реакций гиперчувствительности замедленного типа (образование гранулем) |
| Интерлейкин-1 | Усиление адгезии (повышение экспрессии адгезивных молекул) эндотелиальных клеток, нейтрофилов, эозинофилов, базофилов, моноцитов и (в меньшей степени) лимфоцитов Активация эндотелиоцитов (повышение продукции тромбопластина, ингибитора активации плазминогена, простагландинов, фактора активации тромбоцитов) Стимуляция синтеза острофазных белков (активация гепатоцитов) Пирогенность (прямое действие на гипоталамус) Активация макрофагов (усиление продукции ТНФ-a, интерлейкина-6, фактора активации тромбоцитов, лейкотриенов, тромбоксана А2, простагландинов; аутокринная активация) Синергическое действие с ТНФ-a; повышение чувствительности клеток к ТНФ-a Усиление аккумуляции и активации нейтрофилов Усиление секреции коллагеназы синовиальными клетками; регуляция пролиферации и секреторной активности фибробластов Усиление секреции адренокортикотропного гормона Подавление активности липопротеинлипазы Активация Т-лимфоцитов (продукция лимфокинов); стабилизирующий эффект на пролиферацию В-лимфоцитов и образование антител |
| Интерлейкин-6 | Стимуляция синтеза острофазных белков (активация гепатоцитов) Активация эндотелиоцитов (синтез ростового фактора тромбоцитов, повышение проницаемости) Усиление секреции адренокортикотропного гормона Усиление активации Т- и В-клеток Торможение продукции ТНФ-a при интратрахеальной инсталляции ЛПС Мобилизация нейтрофилов в очаги воспаления |
| Интерлейкин-8 | Хемотаксис нейтрофилов и лимфоцитов Аккумуляция нейтрофилов в очагах воспаления (локальное действие) Подавление эмиграции нейтрофилов в очаги воспаления (системный эффект при внутривенном введении) |
| Гамма- интерферон | Усиление ЛПС-индуцированной секреции макрофагами ТНФ-альфа, интерлейкинов-1, -6; усиление биоцидности макрофагов и экспрессии ТНФ-альфа-рецепторов Усиление экспрессии адгезивных молекул на эндотелиоцитах и лейкоцитах Синергические эффекты с ТНФ-альфа и другими цитокинами Пирогенность (прямое действие на гипоталамус) Усиление мобилизационных реакций нейтрофилов Усиление активации В-лимфоцитов (продукция антител) Усиление адгезии лимфоцитов на эндотелиальных клетках Индукция молекул главного комплекса гистосовместимости классов 1 и 2 (HLA-1, -2) Ослабление продукции гранулоцитарно-макрофагального колониестимулирующего фактора |
Ограниченный набор инструктивных молекул и активированных клеток — лишь начало процесса. При ослаблении гомеостатического контроля число патогенетически значимых сигналов быстро нарастает и отыскать «начало того конца, которым заканчивается начало» невозможно. События эволюционируют по принципу мишень—эффектор, хотя для сетевых (т.е. взаимообусловленных и взаимосвязанных) реакций эти понятия диалектичны, а потому не абсолютны: каждая мишень на определенном этапе выступает в роли эффектора и, наоборот, любой эффектор может оказаться мишенью. Правильнее говорить о первичных, вторичных и т. д. мишенях и эффекторах, имея в виду последовательность их подключения к процессу. Уровень замыкания связей определяет глубину патологии. Чем более простыми средствами устраняются первичный и последующие эффекторы, тем менее сложен комплекс запускаемых ими реакций. Это великолепно иллюстрируется патогенезом септического синдрома. Центральным (инициаторным) звеном здесь является цитокинопосредованное взаимодействие макрофагов и эндотелиальных клеток. Декомпенсированная реакция вынуждает ответ в следующем эшелоне мишеней — эффекторов и т.д. Очевидно, что для обратного развития событий (т.е. для саногенеза) необходим перевес компенсаторных механизмов хотя бы на одной из ступеней патогенетического каскада.
Известно более 100 эндогенных молекул, которые участвуют в воспалении. Только «мудрость тела» способна разобраться в столь сложном переплетении дестабилизирующих сигналов, восстановив над ними контроль. Этим объясняются противоречия в патогенетических трактовках генерализованного воспаления, в частности сепсиса. Есть вопросы и к цитокиновой теории. Непросто, например, обойти наблюдения о повышенном содержании флогогенных цитокинов в крови (включая ТНФ-a) при заболеваниях, не сопровождающихся септическими симптомами. Есть мнение, что более важным является определение цитокиновых рецепторов на клетках-мишенях. С этой точки зрения цитокинемия (т.е. присутствие цитокинов в крови) представляется верхушкой айсберга; патогенетическая сущность феномена остается за кадром. Другой способ избежать противоречий — признать, что модельные опыты с дискретными цитокинами на животных, а тем более in vitro, не отражают реалий тех же молекул при естественной патологии, когда они действуют в сочетаниях — одновременно или последовательно. Это отражено в понятии «сеть цитокинов», где сконцентрирована идея о множественных функциональных контактах инструктивных молекул, взаимном усилении или ослаблении их эффектов и даже качественном изменении конечного результата при воздействии нескольких стимулов. Большое значение имеет реактивность клеток к моменту встречи с активирующими агентами. Некоторые из цитокинов (например, гамма-интерферон) в небольших концентрациях не вызывают уловимых изменений макрофагов, но заметно повышают их чувствительность (праймируют) к другим стимулам. Таким же эффектом обладает и ряд других факторов (фактор активации тромбоцитов, простагландины и пр.), которые участвуют в развитии воспаления. Праймирование широко представлено в реакциях фагоцитов, эндотелиоцитов и других клеток, в том числе при воздействиях, имитирующих септикогенные сигналы.
На протяжении многих лет в терапии сепсиса исходили из примитивного взгляда на его патогенез: процесс связывали исключительно с инвазией бактерий, и лечебная стратегия сводилась к скорейшему применению антибиотиков. Но больные продолжали умирать от респираторной недостаточности, поражения почек, шока. С появлением аппаратуры, имитирующей функции легких, почек, сердца, т.е. всего, на чем базируется современная интенсивная терапия, появилась надежда, что спасение не за горами. Но даже самое активное вмешательство нередко дает лишь временный результат, не справляясь с быстрым развитием процесса.
Сегодня, когда патогенез сепсиса представляется в виде сложнейшей игры эндогенных медиаторов, которая лишь запускается микробным агентом, а затем живет независимо от него, идеология борьбы с септическими осложнениями должна меняться. Новые идеи должны учитывать антицитокиновые эффекты, подавление фактора активации тромбоцитов, оксида азота, эйкозаноидов, комплемента и пр. Продукция цитокинов блокируется, в частности, кортикостероидами, и это может быть одним из механизмов их противовоспалительной активности. Известен опыт с пентоксифиллином, который в высоких концентрациях блокирует транскрипцию ТНФ-a-гена в стимулированных макрофагах. Просматриваются и другие способы выключения генов септикогенных цитокинов, например при помощи антисмысловых нуклеотидов.
Перспективным выглядит подавление адгезивных реакций в системе «эндотелий—лейкоциты». Кроме фармакологических воздействий для этого могут быть использованы антитела против лейкоцитарных и эндотелиоцитарных адгезинов (интегринов, селектинов и пр.), растворимые адгезины и их лиганды. В лечении грамотрицательного сепсиса продолжают возлагать надежды на ЛПС-нейтрализующие антитела, нетоксичные дериваты липида А, блокаду CD14-рецепторов, природные антагонисты ЛПС (в частности, ЛПС-связывающий катионный белок нейтрофилов), растворимые CD14-рецепторы, нацеленные на перехват комплексов ЛПС с белками плазмы.
И все-таки, несмотря на логичность этих идей, они не получили признания в клинике. Попытки внедрить их в терапию септических больных оказались неудачными. Выше уже говорилось о возможных причинах данного явления, в частности отмечалась патогенетическая сложность септического синдрома. Новые поиски должны учитывать патогенетическую поливалентность сепсиса, которая нарастает, как снежный ком, по мере развития реакции. Тактика должна быть комплексной, но концептуально логичной. Наиболее общим является принцип «чем раньше, тем лучше», так как процесс может стать неуправляемым. Это — объективная реальность, и следует быть готовым к разочарованиям, особенно при сравнении клинических результатов с экспериментальными моделями, где события обычно не заходят столь далеко. Всегда следует ожидать худшего, памятуя о том, что опережающая диагностика — основа успешной терапии сепсиса. Ориентиром должны быть малые (формально не угрожающие) признаки, когда порочный круг (точнее сеть порочных кругов) еще не сформировался и развитие процесса можно остановить. К счастью, организм сам нередко справляется с этой задачей, обрывая трагическую цепь событий на одном из этапов. Нет смысла перечислять все вопросы и противоречия, которые ждут своего разрешения. Они позволяют воздержаться от чрезмерных увлечений, выдвигая задачи для новых исследований.
Антибиотики
Жизнь убивает жизнь. Л. Пастер Выбор антимикробного препарата прост, пока не требуется его безвредность. В. Ослер
- Антибиотики и биологический антагонизм.
- Принцип «волшебной пули».
- Природные антибиотики и химические вариации.
- Ингибиторы синтеза клеточной стенки бактерий.
- Ингибиторы синтеза белка.
- Ингибиторы плазматической мембраны.
- Резистентность бактерий к антибиотикам: тактика и стратегия.
- Дилемма выбора.
В широком смысле любое проявление антагонизма между живыми объектами есть антибиоз. Именно так представлял антибиоз Ж. Вильмен, предложивший этот термин в 1889 г. Понятие было забыто и возродилось в начале 1940-х гг. в связи с практическим использованием антагонизма между микробами.
Антибиотиками было предложено называть продукты микроорганизмов, которые в ничтожных концентрациях убивают или задерживают размножение других микробов. В такой формулировке (ее ввел С. Ваксман в 1941—1942 гг.) антибиотики легко отделимы от антибактериальных веществ иного (немикробного) происхождения. Это определение сохраняет смысл и сегодня, когда большинство антибиотиков синтезируется или усовершенствуется in vitro. Химические модификации нацелены на улучшение того, что создала природа.
Вместе с тем развитие антибактериальной терапии привело к сглаживанию различий между природными и искусственными препаратами. Некоторые наименования последних имитируют антибиотики, внося путаницу в номенклатуру: норфлоксацин, цитрофлоксацин и др. Это побуждает отказаться от первоначальной идеи относительно термина «антибиотики», заставляя использовать его для обозначения различных лечебных продуктов с антимикробным эффектом. Этого правила мы будем придерживаться и в нашем очерке, считая, что понятие «антибиотик» во многом утратило свой классический смысл.
Антагонизм в мире микробов — весьма распространенное явление, которое известно давно, с момента зарождения микробиологии. Отсюда не удивительно, что среди первых, кто обратил на это внимание, был Л. Пастер. В 1877 г. он вместе с Жубером отметил, что заражение животных сибирской язвой можно предотвратить, если вместе с культурой сибиреязвенной палочки ввести другие бактерии. Пастер и Жубер предсказали микробному антибиозу блестящее будущее. Еще раньше, в 1871—1872 гг., российские ученые В.А. Манассеин и А.Г. Полотебнов наблюдали эффект при лечении зараженных ран прикладыванием плесени. В начале XX столетия был популярен препарат из синегнойной палочки — пиоционаза. Его применяли для лечения бактериальных инфекций, хотя, возможно, это курьез, не имеющий отношения к истории этиотропной терапии (см. «Синегнойная палочка»).
В 1929 г. А. Флеминг обнаружил штамм пеницилл (Penicillium notatum), продукты которого обладали выраженной активностью против грамположительных бактерий. Потребовалось десять лет (тогда все находились под впечатлением только что открытых сульфаниламидов), чтобы пенициллином серьезно заинтересовались биохимики. В 1940 г. Х. Флори и Э. Чейн получили стабильный препарат очищенного пенициллина — первый антибиотик, нашедший широкое применение в клинике (вместе с Флемингом они отмечены Нобелевской премией). Правда за год до этого Р. Дюбо выделил из Bacillus brevis тиротрицин (более известен один из его компонентов — грамицидин), но из-за токсичности он не вызвал большого энтузиазма.
С тех пор открыты и синтезированы десятки тысяч антибиотиков, но лишь немногие из них оказались пригодными для клинического использования, сочетая высокую антибактериальную активность с необходимыми фармакологическими параметрами, прежде всего с безвредностью. В этом принцип «волшебной пули» П. Эрлиха — убить живое в живом, не вредя живому, т.е. уничтожить паразита, не нанося ущерба хозяину. Впрочем, П. Эрлиху принадлежит скорее формулировка, нежели сама идея. Мысль об этиотропной терапии возникла гораздо раньше — к ней логично приходили те, кто верил в contagium vivum — живое начало заразных болезней. Так, еще в XVI в. Д. Фракасторо считал, что при лечении врач должен стремиться убить в организме контагий, а если это невозможно, то по крайней мере ослабить или изгнать его. Призыву Фракасторо эмпирически следовали при лечении ряда заболеваний, например малярии (хина) и сифилиса (препараты ртути). В 1908 г. П. Эрлих на основе органических соединений мышьяка синтезировал сальварсан (лат. спасающий) — знаменитый препарат для лечения сифилиса. Открытие окрылило, но надежды на химиотерапию других инфекций стали рушиться. В конце концов сам Эрлих заявил, что случай с сальварсаном и сифилисом — исключение. Спирохеты — особые микробы, они крупнее типичных бактерий и отличаются от них по своим свойствам. Борьба с другими бактериями при помощи «волшебных пуль» бесперспективна.


К счастью, П. Эрлих оказался неправ. Все бактерии располагают жизненно важными механизмами, которые отсутствуют (или сильно отличаются) у эукариотических клеток. Поэтому они могут быть избирательно атакованы, погибать (бактерицидный эффект) или прекращать размножаться (бактериостатический эффект) в присутствии определенных агентов (рис. 1). Бактерицидность надежнее, хотя и бактериостатичность обычно действенна, так как бактерии, находящиеся под контролем, элиминируются факторами самозащиты хозяина.Первым, но существенным опровержением грустного прогноза Эрлиха явилось лечение бактериальных инфекций сульфаниламидами. Начало положил Г. Домагк, предложивший в 1935 г. пронтозил (красный стрептоцид). Его действующим началом оказался сульфаниламид, который высвобождается при разложении пронтозила в организме (рис. 2). Селективное (бактериостатическое) действие сульфаниламида и его многочисленных производных объясняется конкурентной блокадой синтеза фолиевой (точнее тетрагидрофолиевой) кислоты — предшественника пуриновых и пиримидиновых оснований. Клетки высших организмов и некоторых бактерий ауксотрофны по фолиевой кислоте, т.е. не синтезируют ее и должны получать в готовом виде. Поэтому сульфаниламиды для них не опасны.

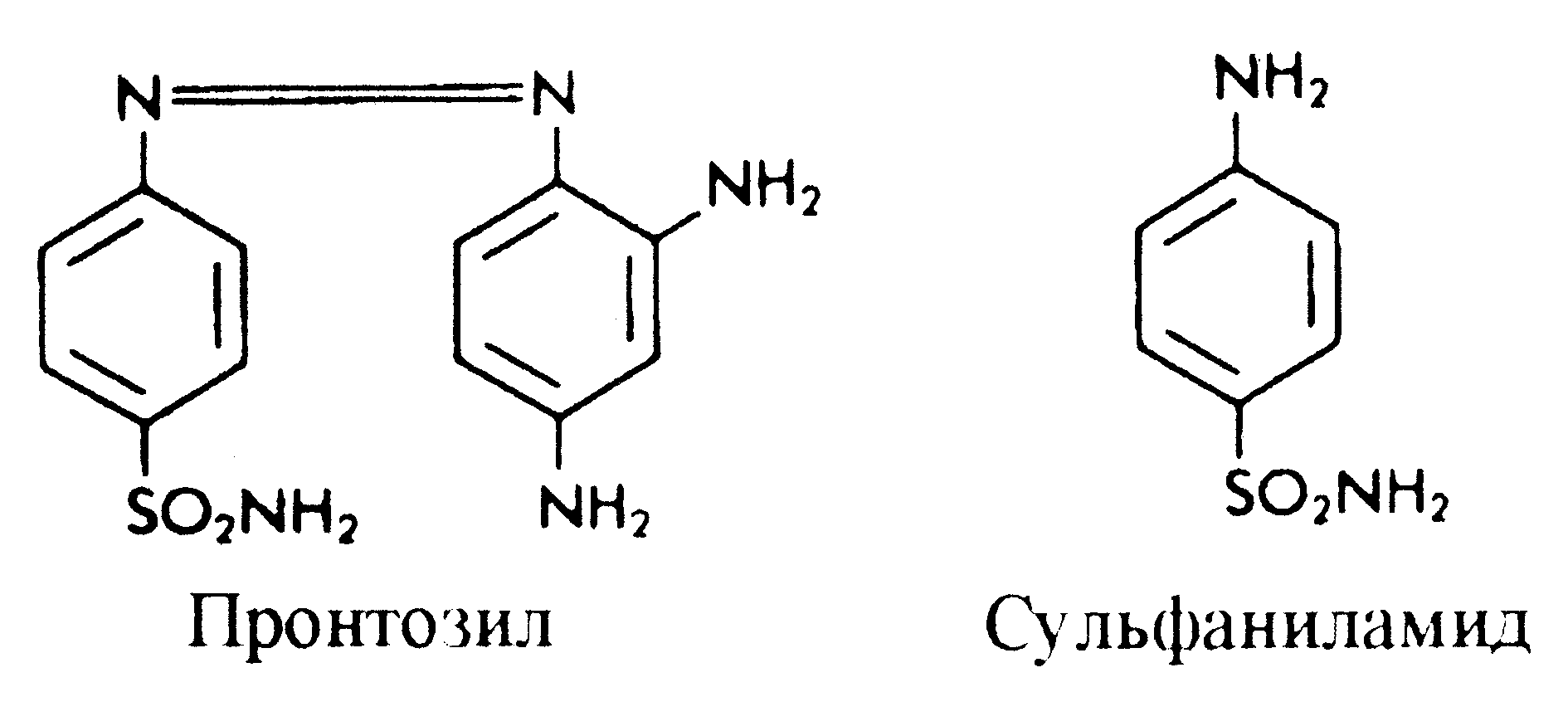

Успех сульфаниламидов едва не сыграл роковую роль в истории антибактериальной терапии. По крайней мере Флеминг, разочарованный неудачными попытками очистить пенициллин, прекратил работу в этом направлении. К счастью, он сохранил штамм плесени, подаренный ему и всему человечеству случаем (рис. 3). Именно это определило будущее химиотерапии. По действию на пиогенные бактерии пенициллин значительно превосходил сульфаниламиды.
Антибиотики (антимикробные препараты) принято классифицировать по источнику получения, химической структуре, спектру антимикробной активности и механизму действия (см. таблицу). Список антибиотиков весьма обширен, в чем повинно не только разнообразие синтетических производных, но и изобилие терминов, которые присваиваются однотипным препаратам разными фирмами. Например, пенициллин G (бензилпенициллин) имеет более 20 синонимов, ампициллин — около 60 и т.д. Это затрудняет ориентирование в море антимикробных средств, требуя понимания микробиологических и фармакологических принципов этиотропного лечения, достоинств и ограничений каждого препарата, причин возможных неудач и осложнений.
Таблица 1
Основные типы антибактериальных препаратов (антибиотиков)
| Семейство | Класс | Примеры | Механизм действия |
| 1. Бета-лактамы | Пенициллины | Блокада синтеза клеточной стенки (пептидогликана) | |
| Природные | Пенициллин G Пенициллин V | ||
| Полусинтетические: | |||
| Аминопенициллины | Ампициллин | ||
| Карбоксипенициллины | Карбенициллин Карфециллин | ||
| Пенициллиназо- резистентные | Метициллин Клоксациллин | ||
| Уреидопенициллины | Азлоциллин Мезлоциллин Пиперациллин | ||
| Цефалоспорины | |||
| Природные | Цефалоспорин С | ||
| Полусинтетические: | |||
| 1-е поколение | Цефалотин Цефазолин | ||
| 2-е поколение | Цефамандол Цефуроксим | ||
| 3-е поколение | Цефотаксим Цефтазидим | ||
| Цефамицины* | Цефокситин | ||
| Карбапенемы | Имипенем | ||
| Монобактамы | Азтреонам | ||
| 2. Гликопептиды | Ванкомицин Тейкопланин | Блокада синтеза клеточной стенки (пептидогликана) | |
| 3. Циклосерин | Блокада синтеза клеточной стенки (пептидогликана) | ||
| 4. Аминогликозиды | Стрептомицин Неомицин Канамицин Гентамицин | Блокада синтеза белка | |
| Новое поколение | Тобрамицин Амикацин Эритромицин | ||
| 5. Макролиды | Олеандомицин | Блокада синтеза белка | |
| Новое поколение | Азитромицин Кларитромицин Рокситромицин | ||
| 6. Линкозамиды | Линкомицин Клиндамицин | Блокада синтеза белка | |
| 7. Тетрациклины | Тетрациклин Окситетрациклин Доксициклин | Блокада синтеза белка | |
| 8. Хлорамфеникол (левомицетин) | Блокада синтеза белка | ||
| 9. Фузидиновая кислота (фузидин) | Блокада синтеза белка | ||
| 10. Стрептограмины | Синекрид | Блокада синтеза белка | |
| 11. Оксазолидиноны | Зивокс | Блокада синтеза белка | |
| 12. Рифамицины | Рифампин | Блокада синтеза м-РНК | |
| 13. Циклические полипептиды | Полимиксины | Повреждение плазматической мембраны | |
| 14. Сульфаниламиды/ диаминопиримидины | Подавление синтеза ДНК | ||
| 15. Производные нитромидазола | Метронидазол | Повреждение ДНК | |
| 16. Производные нитрофурана | Фуразолидон Фурагин Нитрофурантон | Повреждение ДНК (?!) | |
| 17. Фторхинолоны | Норфлоксацин Офлоксацин Левофлоксацин | Инактивация ДНК-гиразы |
*Иногда о них говорят как о цефалоспоринах 4-го поколения.
Основные вопросы, которые следует задавать, применяя антибиотик, сводятся к следующему:
- что он из себя представляет (химическая структура, природный или синтетический продукт);
- как он действует (мишень, механизм действия);
- фармакодинамика (всасывание, распределение в тканях организма, метаболизм и экскреция) и отсюда предпочтительный способ применения;
- когда его следует применять (спектр активности и главные клинические ситуации);
- возможные проблемы (токсичность, микробная резистентность);
- стоимость (значительные колебания цен позволяют выбрать близкий, но более дешевый препарат).
Мы остановимся лишь на микробиологических аспектах проблемы, связанных с механизмом действия антибиотиков, лекарственной резистентностью бактерий, и принципах ее преодоления.
Сегодня широко практикуется эрлиховский принцип химических вариаций, когда, нащупав рациональную основу, искусственно синтезируется множество новых антибиотиков, из которых отбирают удачные экземпляры. Особенно богатым источником природного сырья оказались актиномицеты, прежде всего стрептомицеты. Это «атипичные» бактерии, которые имеют формальное сходство с плесневыми грибами, образуя мицелиальные формы. Стрептомицеты являются высшими актиномицетами, так как, подобно грибам, размножаются спорами.
Классификация по химическим свойствам основана на базисном сходстве молекул. Иногда его непросто заметить (по крайней мере, не химику-профессионалу) в изобилии второстепенных деталей, от которых зависит клиническая ценность препарата. Практически важным является представление о преимущественной направленности, широте и ограниченности антимикробного эффекта антибиотических препаратов, их фармакодинамических параметрах. Большой интерес вызывают механизмы, определяющие активность антибиотиков, молекулярные мишени, на которые направлено их действие, и способы, благодаря которым бактерии избегают гибели.
Механизмы антибактериального эффекта
Общим для всех антибиотиков является то, что они влияют на бактерии в фазе активного роста и размножения. Это связано с тем, что антибиотики вмешиваются в метаболизм бактериальных клеток, обычно не повреждая готовые структуры покоящихся бактерий. Этим можно объяснить их опережающее воздействие на патогенные бактерии при относительно слабом влиянии на нормальную микрофлору. Патогенетически значимая колонизация требует от возбудителей быстрого размножения и усиленной метаболической активности. Это повышает уязвимость бактерий для антимикробных агентов. Лишь длительное применение антибиотиков, поддерживающее их концентрацию в организме, создает угрозу малоактивным бактериям.

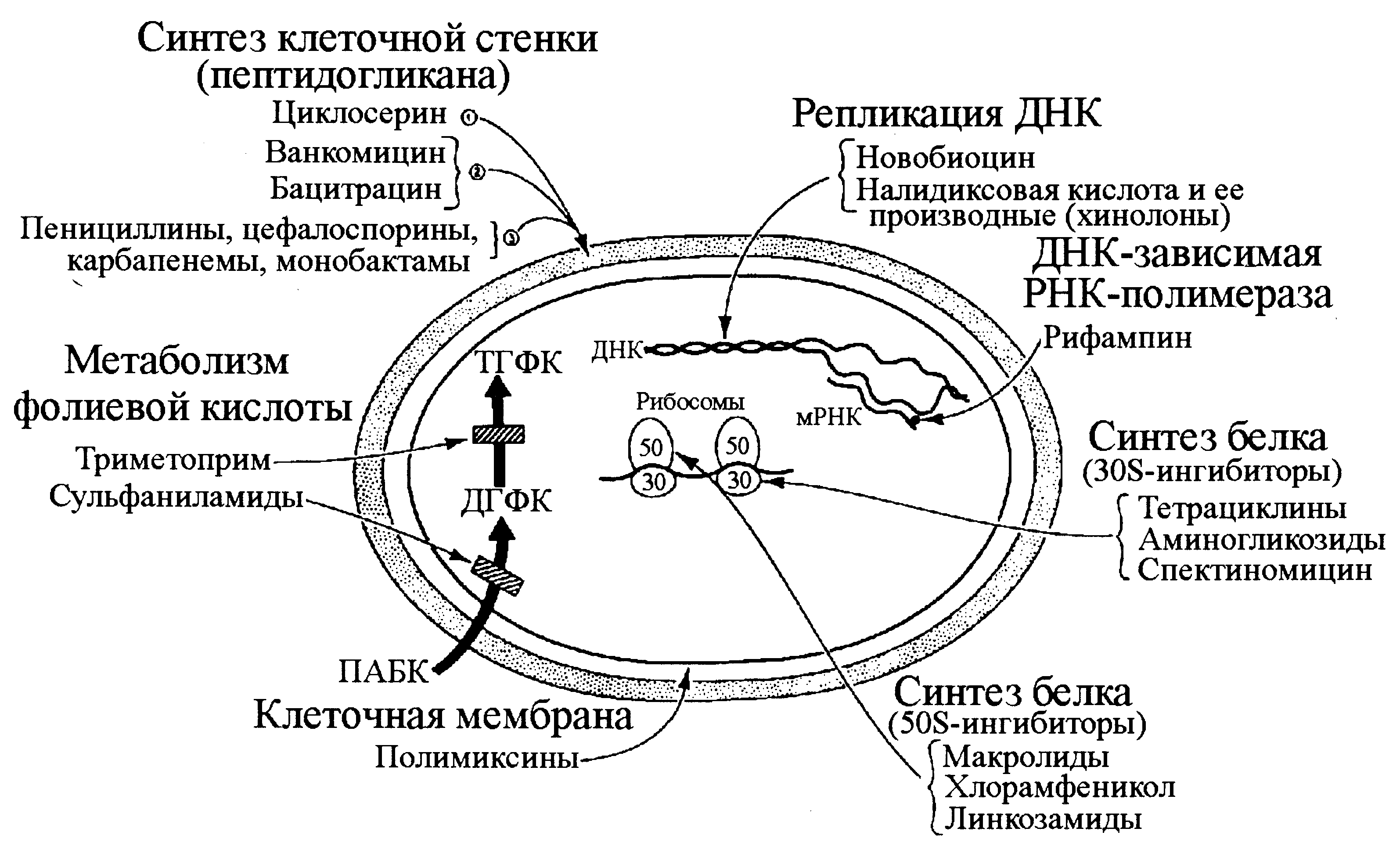
ДГФК — дигидрофолиевая кислота, ТГФК — тетрагидрофолиевая кислота
Отсюда следует практически важный вывод: антибиотики при хронических инфекциях менее эффективны, чем при острых, и едва ли полезны при бессимптомном и малосимптомном носительстве болезнетворных бактерий, репликативная активность которых в подобных случаях не отличается от нормофлоры.
Основные точки действия антибактериальных агентов проиллюстрированы на рис. 4.
Ингибиторы синтеза клеточной стенки бактерий (антипептидогликановые антибиотики)
Клеточная стенка бактерий — идеальная мишень для высокоизбирательного антибактериального эффекта, так как ее структурная основа, пептидогликан (или муреин, от лат. murus — стенка), уникальна и жизненно необходима для прокариотов. Эукариотические клетки лишены не только пептидогликана, но и всей атрибутики, необходимой для его синтеза.
Пептидогликан определяет механическую прочность клеточной стенки, позволяя выдерживать высокое осмотическое давление бактериальной цитоплазмы. Это гетерополимер, построенный из параллельных полисахаридных (гликановых) цепей, которые состоят из чередующихся аминосахаров (N-ацетилглюкозамин и N-ацетилмурамовая кислота), однотипных боковых тетрапептидов, связанных с N-ацетилмурамовой кислотой, и межпептидных мостиков, состоящих из нескольких аминокислот. Межпептидные мостики сшивают пептидогликановые цепи, превращая их в единую многослойную макромолекулу (рис. 5), которая непрерывным слоем окружает протопласт (рис. 6).

Рис. 6. Электронограммы клеток (а) и пептидогликана (б) золотистого стафилококка
У грамотрицательных бактерий боковые тетрапептиды соединены напрямую пептидной связью, оставляя концевую аминокислоту (d-аланин) для стыковки (через липопротеин) с наружной мембраной (рис. 7). Все пептидогликаны построены по общей схеме с видовыми вариациями в пептидных (транспептидных) субъединицах.

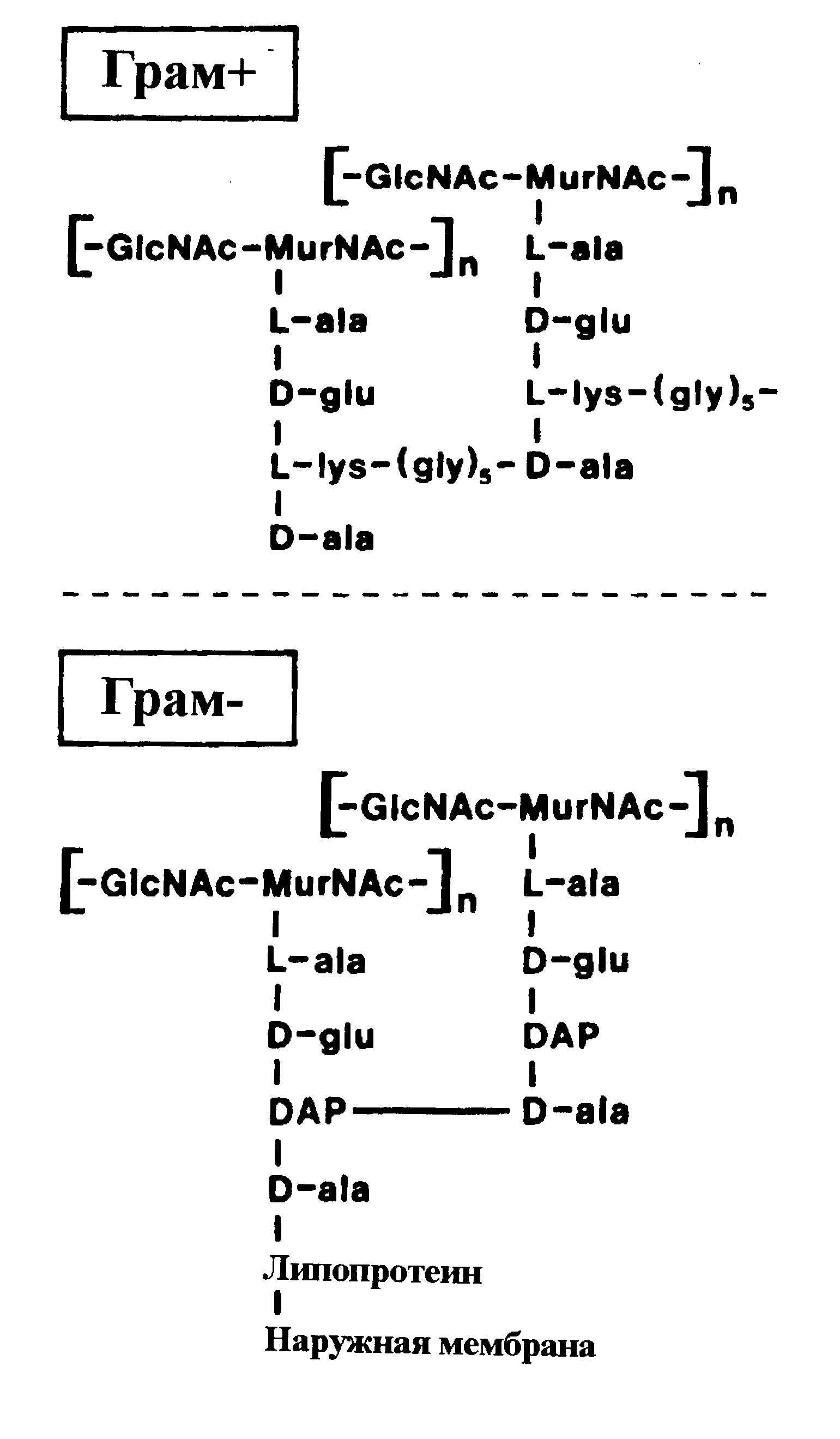
протеином, который соединен с наружной мембраной

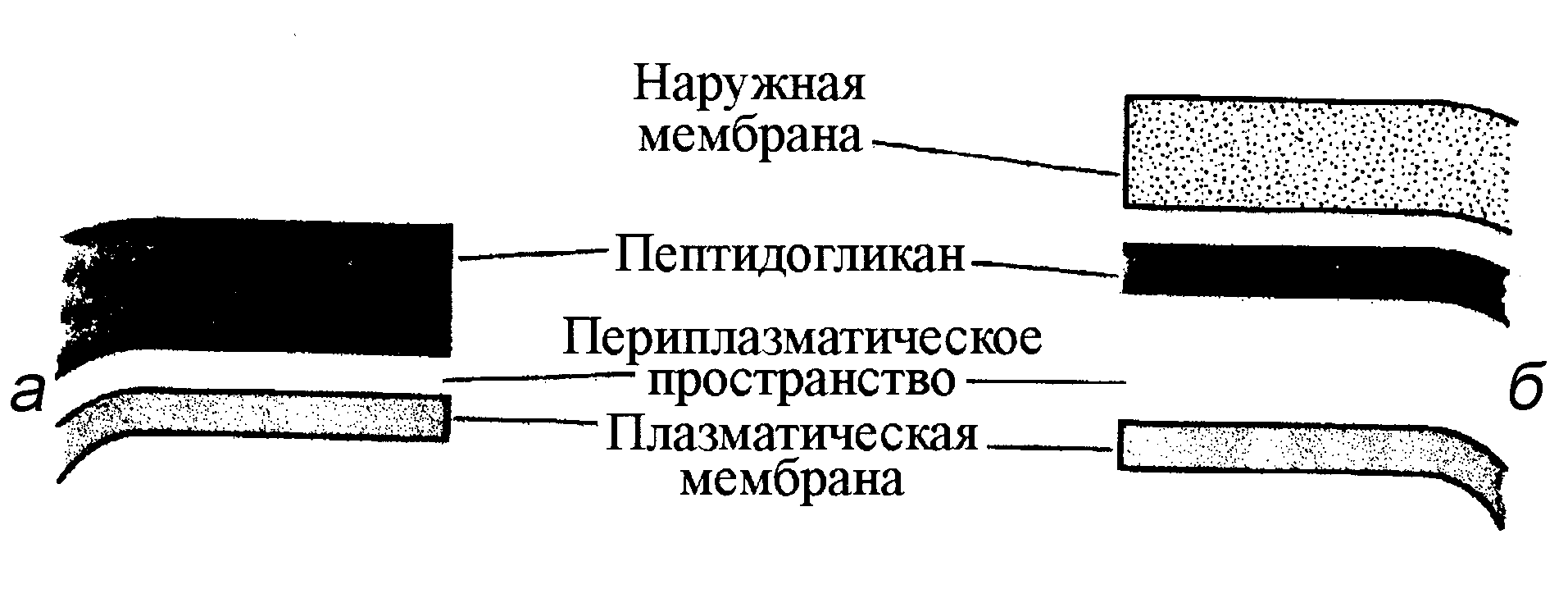
Пептидогликан есть почти у всех бактерий. Важными в медицинском отношении исключениями являются микоплазмы и хламидии. Это означает, что они не чувствительны к антибиотикам, блокирующим синтез клеточной стенки.
Особенно мощный пептидогликановый каркас имеют грамположительные бактерии. У грамотрицательных бактерий он много тоньше, хотя это компенсируется наличием наружной мембраны, которая, выполняя ряд важных метаболических и патогенетических функций, служит дополнительной защитой, в том числе от антибиотиков (рис. 8).
Синтез пептидогликана включает три базисных этапа:
1) образование (в цитоплазме) гликопептидных субъединиц и их транспорт в цитоплазматическую мембрану;
2) объединение субъединиц в гликопептидные цепи (это происходит в плазматической мембране);
3) сшивание пептидогликановых цепей межпептидными связями с образованием пептидогликанового каркаса.
Этот этап совершается на внешней поверхности плазматической мембраны под влиянием ферментов — транспептидаз. Они известны как пенициллинсвязывающие белки, потому что служат мишенью для бета-лактамных антибиотиков.
Антипептидогликановые антибиотики действуют бактерицидно, вызывая лизис бактерий. Но они лишь запускают процесс: завершение обеспечивают собственные ферменты бактерий — аутолизины. Их физиологическая функция состоит в контролируемом расщеплении пептидогликана при делении бактериальных клеток. Антибиотики неизвестным способом снимают этот контроль, провоцируя саморазрушение клеточной стенки и разрыв плазматической мембраны. Без такой поддержки антибиотики лишь блокируют синтез пептидогликана. В этом их принципиальное отличие от антипептидогликановых ферментов — мурамидаз, действующих по типу лизоцима. Бактериальные аутолизины принадлежат к этой категории.
Почти все антибиотики, блокирующие синтез клеточной стенки, относятся к семейству бета-лактамов (см. таблицу). Их структурная и функциональная основа представлена бета-лактамным кольцом (рис. 9), которое связывается с транспептидазами, препятствуя завершению синтеза пептидогликанового каркаса. Наиболее важными бета-лактамами являются пенициллины и цефалоспорины. Они представлены множеством структурных модификаций природных субстратов — пенициллина (продукт гриба рода Penicillium) и цефалоспорина (грибы рода Cephalosporium). Впрочем, и другие бета-лактамы (особенно карбапенемы и монобактамы) зарекомендовали себя как перспективные антибиотики и служат предметом активного применения.

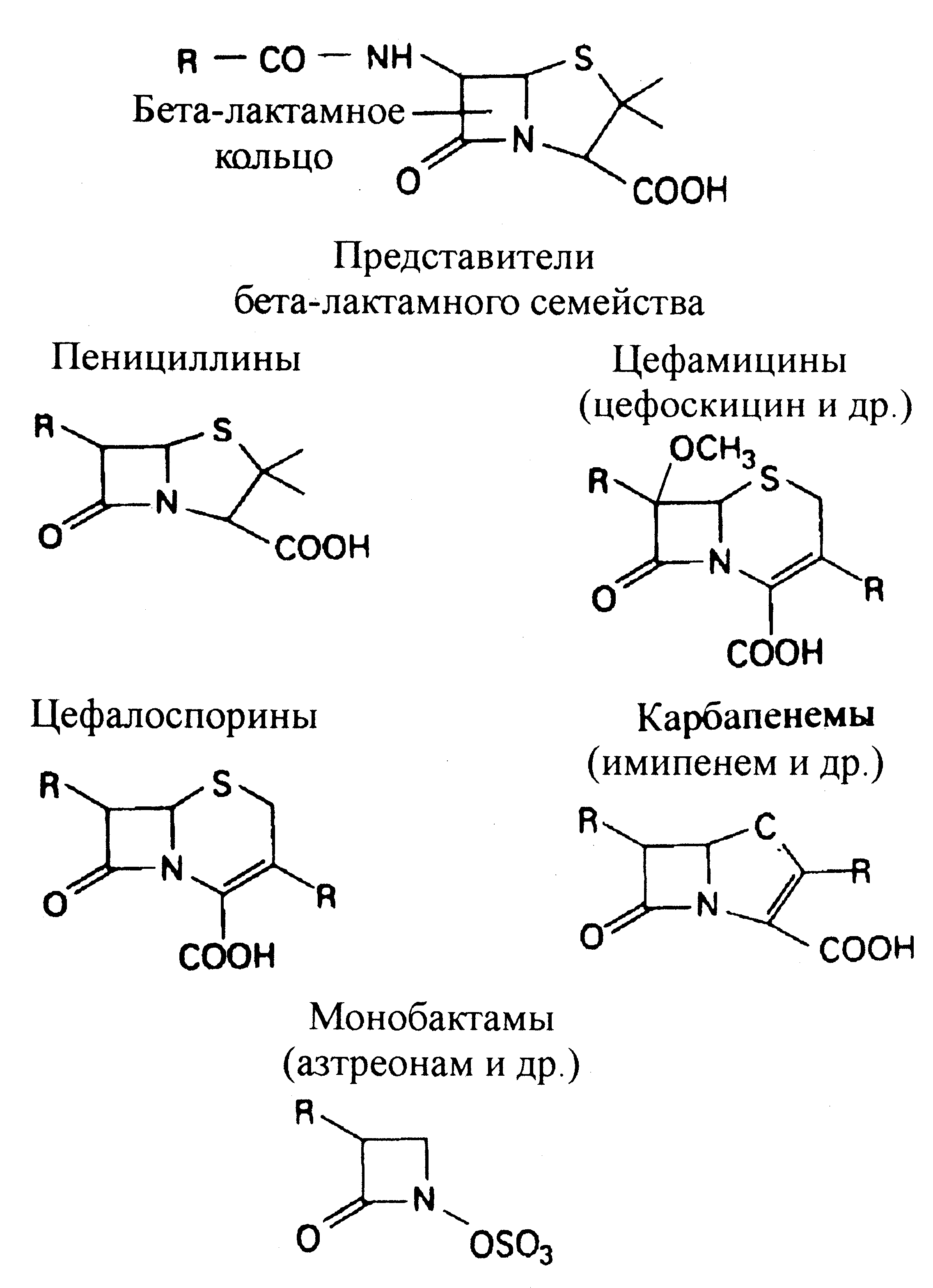
Кроме бета-лактамов, синтез клеточной стенки блокируют гликопептидные антибиотики (ванкомицин, тейкопланин), а также бацитрацин и циклосерин. Они действуют на более ранних этапах синтеза пептидогликана, подавляя образование гликопептидных субъединиц (циклосерин) и их полимеризацию в гликопептидные цепи (ванкомицин, бацитрацин).
Ингибиторы синтеза белка (антирибосомальные антибиотики)
Синтез белка, в основе которого лежит трансляция мРНК на рибосомах — многоступенчатый процесс, где задействовано множество ферментов и структурных субъединиц. Поэтому не случайно, что такая сложная система является мишенью для антибиотиков. По ряду признаков белоксинтезирующий аппарат прокариот отличается от рибосом эукариотических клеток. Бактериальные рибосомы состоят из меньших по размеру субъединиц (30S и 50S — прокариоты, 40S и 60S — эукариоты), более мелких молекул РНК и включают меньше белков. Принципиально, что эти различия находят качественное выражение, определяя селективность мишеней рибосомального цикла для антибактериальных воздействий (рис. 10). Аминогликозиды и тетрациклины связываются с 30S-субъединицей, нарушая образование инициаторного рибосомального комплекса. Тетрациклины обладают бактериостатическим эффектом, обратимо блокируя присоединение к рибосомам инициаторной аминоацил-транспортной РНК (формилметионил-тРНК).

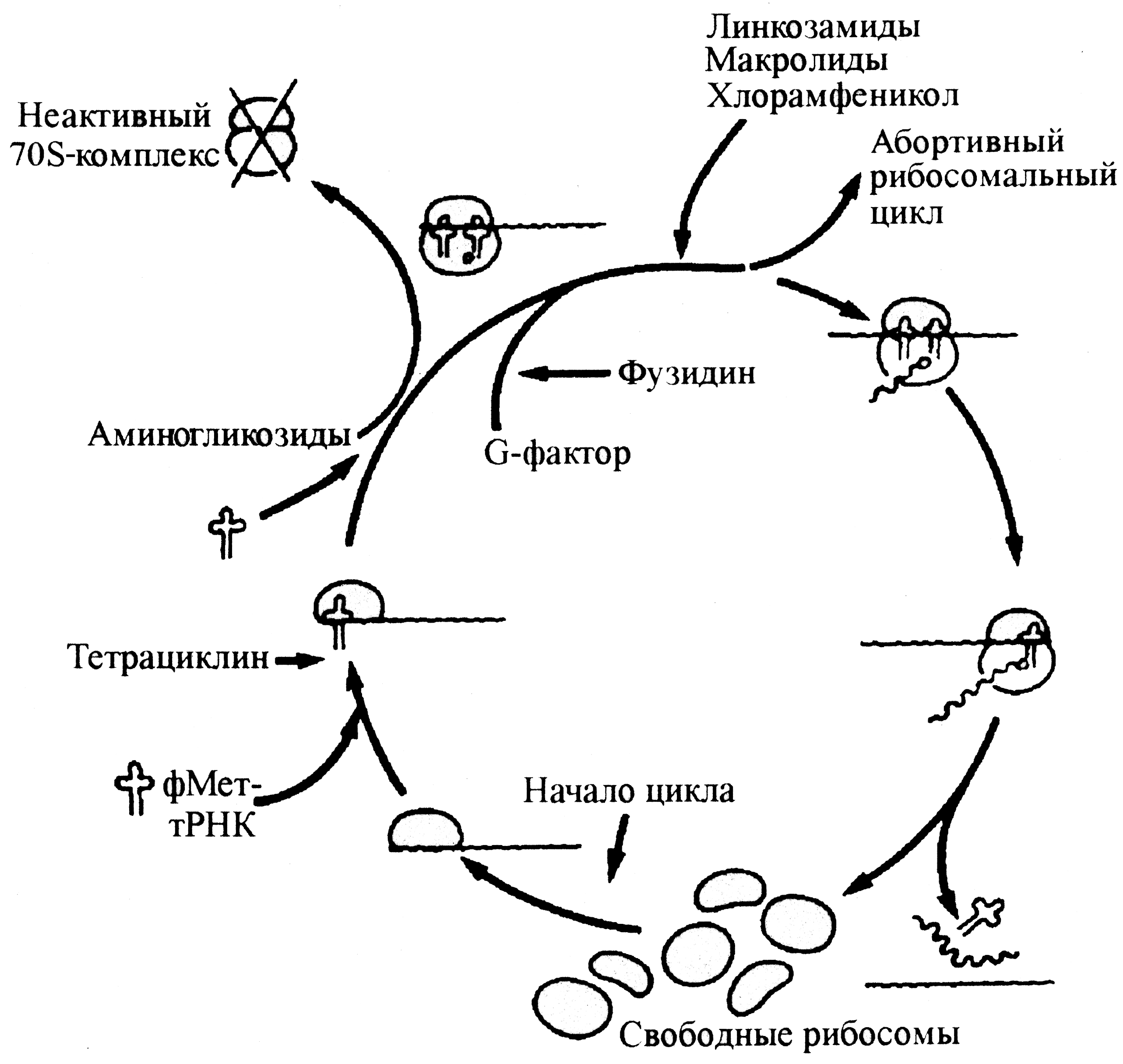
цикл)
Аминогликозиды убивают бактерии, вызывая образование функционально непригодных (неспособных к диссоциации—реассоциации) 70S-рибосом, а также извращая считывание информации с матричной РНК (мРНК). Макролиды, хлорамфеникол, линкомицин/клиндамицин соединяются с 50S-субъединицей вблизи сайта, фиксирующего аминоацил-тРНК. Это обрывает элонгацию (удлинение) пептидных цепей. В отличие от аминогликозидов и тетрациклинов, блокирующих синтез белка в момент его зарождения, здесь прерывается уже начавшийся цикл. После удаления антибиотика процесс возобновляется, т.е. эффект бактериостатичен. К такому же финалу ведет фузидиновая кислота (фузидин), но механизм здесь иной — инактивация фермента (транслоказы, или фактора элонгации G), который обеспечивает передвижение рибосомы по мРНК, создавая условия для включения в пептидную цепь очередной аминокислоты.
Ингибиторы синтеза нуклеиновых кислот
Дискриминация нуклеиновых кислот достигается тремя способами: 1) блокада синтеза предшествеников пурин-пиримидиновых оснований (сульфаниламиды), 2) подавление репликации ДНК (хинолоны, метронидазол) и 3) инактивация РНК-полимеразы (рифамицины).
Клиническим лидером рифамицинов является рифампицин (рифампин). Он связывается с РНК-полимеразой, блокируя транскрипцию, т.е. синтез матричных РНК. Избирательная токсичность основана на том, что бактериальные полимеразы по аффинности к рифамицинам превосходят аналогичные ферменты высших клеток.
Действие метронидазола проявляется после восстановления его нитрогруппы бактериальными белками — ферродоксинами. Это превращает метронидазол в мутаген, способный взаимодействовать и повреждать ДНК бактерий. Ферродоксины имеются у облигатных анаэробов и микроаэрофилов, но отсутствуют в аэробах и факультативных анаэробах. На них метронидазол не действует.
Большой интерес вызывают фторхинолоны. Они (так же, как сульфаниламиды и метронидазол) не относятся к «истинным» антибиотикам, являясь продуктами, синтезированными на основе налидиксовой кислоты. Эффект хинолонов связан с инактивацией ДНК-гиразы — фермента, обеспечивающего суперспирализацию (упаковку) бактериальной хромосомы. Без этого ДНК не помещается в клетках, что делает бактерии нежизнеспособными. Сходные ферменты (топоизомеразы) высших животных не чувствительны к хинолонам.
Ингибиторы цитоплазматической мембраны
Число антибиотиков, специфически действующих на мембраны бактерий, невелико. Наиболее известны тиротрицин (смесь грамицидина и тироцидина) и полимиксины — циклические полипептиды, к которым чувствительны только грамотрицательные бактерии. Они лизируют клетки, действуя как катионные детергенты, т.е. повреждая фосфолипидный матрикс клеточных мембран. Их избирательность значительно уступает другим антибиотикам, так как мембраны бактерий и высших клеток имеют много общего. Из-за токсичности они применялись лишь для лечения местных процессов и не вводились парентерально. В настоящее время не применяются.
Мембранотропностью обладают полиеновые антибиотики (амфотерицин В, нистатин, леворин), которые используются в терапии грибковых заболеваний. Они повреждают мембраны, связываясь со стерольными группировками. Бактерии не чувствительны к полиенам, так как за исключением микоплазм лишены стеролов. Этого нельзя сказать о клетках высших животных, поэтому отыскать специфические мишени для антигрибковых препаратов весьма непросто. К счастью, и здесь удалось «зацепиться»: в отличие от млекопитающих, у которых доминирует холестерол, плазматическая мембрана грибов содержит его химический родственник — эргостерол. В этой связи показательны наблюдения о возможности избирательной блокады стерольного синтеза у грибов. Этого удалось добиться при помощи азольных производных, которые ингибируют один из ключевых ферментов биосинтеза эргостерола — цитохром Р450-деметилазу (ланостерол С14-деметилазу). Такой же фермент есть у человека и других млекопитающих. Он обеспечивает образование холестерола и менее чувствителен к соединениям группы азолов. Кроме средств для наружного применения (клотримазол, миконазол и др.), на этой основе разработаны препараты для лечения системных микозов — кетоконазол и флюконазол. Последний особенно успешно используется в лечении кандидозных инфекций. Это еще один пример, когда на помощь естественным антибиотикам приходят синтетические продукты, не известные живой природе.
Резистентность бактерий к антибиотикам
Бытует легенда, что, комментируя восторг по поводу успешного применения пронтозила (родоначальника сульфаниламидов), И.П. Павлов говорил, что, действуя на микробы, следует помнить об их собственных интересах. Главный интерес — не оригинален. Как и у всего живого — это стремление к выживанию, т.е. к закреплению себя в биосфере. Фраза оказалась пророческой. Возможности бактерий к развитию устойчивости против антибактериальных агентов превзошли многие пессимистические прогнозы. Вслед за стафилококком волна адаптивных перестроек захватила и другие бактерии, сделав проблему универсальной. Резистентность и ее преодоление стали по сути самостоятельным разделом в учении об антибиотиках и антимикробной терапии, побуждая непрерывно трудиться над разрешением этого «реприманда природы».
В медицинском смысле резистентными следует считать бактерии, если они не обезвреживаются такими концентрациями агента, которые возникают в организме при введении фармакологических, т.е. клинически реальных дозировок. Опасная тенденция нарастания устойчивости к антибиотикам поддерживается генами резистентности (r-генами) и условиями, способствующими их распространению. В считанные десятилетия (даже годы) лекарственная резистентность превратилась в тревожную клинико-эпидемиологическую проблему, значение которой особенно велико для внутрибольничных инфекций. Одним из характерных признаков так называемых госпитальных штаммов является множественная устойчивость к антибиотикам, которая возникает в результате широкого обмена r-генами и селекции резистентных штаммов на фоне обильного применения антибактериальных препаратов.
Понять природу лекарственной устойчивости бактерий помогает представление об общих принципах реализации антимикробного эффекта. Они сводятся к следующему: агент 1) должен связаться с бактериями и пройти через их оболочку; 2) должен быть доставлен к месту действия и 3) вступить во взаимодействие с внутриклеточными мишенями. Формирование устойчивости возможно на каждом из этих этапов. Бактерии действуют, как квалифицированные биохимики, изобретая и совершенствуя способы борьбы против нацеленной на них агрессии.
Стратегия резистентности базируется на следующих механизмах:
- снижение проницаемости клеточной стенки для антимикробного агента и подавление его транспорта к внутриклеточным мишеням;
- ускоренное выделение агента из клетки;
- модификация мишеней;
- снижение физиологической значимости мишени за счет дублирования способов образования жизненно важных метаболитов;
- конкурентное связывание (перехват) антимикробного агента;
- инактивация агента.
Сначала очень кратко остановимся на генетическом аппарате прокариот. Он представлен единственной хромосомой, где находится большинство (около 3000) генов, и миниатюрными ДНК-молекулами — плазмидами. Самые крупные из них включают не более 200 генов, но тем не менее очень важны для своих хозяев. Это прежде всего связано с тем, что плазмиды несут гены адаптации, которые повышают экологическую пластичность бактерий, позволяя приспосабливаться к необычным условиям и выживать в экстремальных ситуациях. Многие из таких генов относятся к транспозонам — подвижным (прыгающим) генам. Они охотно переходят из репликона в репликон (например, из плазмиды в плазмиду, из хромосомы в плазмиду и т.д.) и вкупе с мобильностью самих плазмид обеспечивают быстрое распространение генов внутри популяций, видов и даже между разными видами бактерий.
Резистентность бывает первичной и приобретенной, т.е. предсуществует до встречи с антимикробным агентом или возникает после взаимодействия с ним. Первичная резистентность закодирована в хромосомных генах и является видовым признаком. Например, грамотрицательные бактерии (за исключением нейссерий) малочувствительны к природным пенициллинам, так как они плохо проникают через их наружную мембрану. У синегнойной палочки этому способствует барьер из слизи, непроницаемой для многих агентов. Бактерии могут быть лишены мишени, атакуемой антибиотиком, или факторов, которые обеспечивают их активацию. Так, микоплазмы не имеют клеточной стенки и поэтому не чувствительны к бета-лактамам. Обладая высокой активностью против аэробов и факультативных анаэробов, аминогликозиды не действуют на анаэробные бактерии, которые лишены транспортных систем, необходимых для ассимиляции аминогликозидных антибиотиков. Напротив, метронидазол эффективен против облигатных анаэробов, но бессилен против аэробных бактерий. Дело в том, что для активации метронидазола требуется восстановление его нитрогруппы, а такой способностью обладают только анаэробы.
Приобретенная (вторичная) резистентность непредсказуема, а потому доставляет особенно много хлопот. Ее основным механизмом является приобретение генов резистентности (r-генов), переносимых транспозонами и плазмидами. Плазмида, содержащая r-гены, называется R-плазмидой, или R-фактором. R-плазмиды могут иметь единственный r-ген или быть носителем r-генов к нескольким антибиотикам. Полирезистентность возникает и в том случае, если бактериальная клетка содержит несколько разных R-плазмид.
Важно понимать, что антибиотики не индуцируют образование r-генов, а лишь способствуют их отбору. Такие гены обнаружены у штаммов, которые были законсервированы задолго до внедрения антибиотиков, а также у изолятов из регионов, где данный антибиотик никогда не применялся. В подобных случаях r-гены обнаруживаются в единичных бактериях (точнее клонах), так как, не создавая преимуществ своим обладателям, не получают распространения. Антибиотик действует как селекционирующий фактор. В его присутствии выживают лишь резистентные клоны, из которых формируется устойчивая популяция — вплоть до устойчивого вида, как это случилось для пенициллинрезистентных стафилококков (резистентностью к природным пенициллинам обладают 90—98% циркулирующих штаммов S. aureus) (рис. 11).

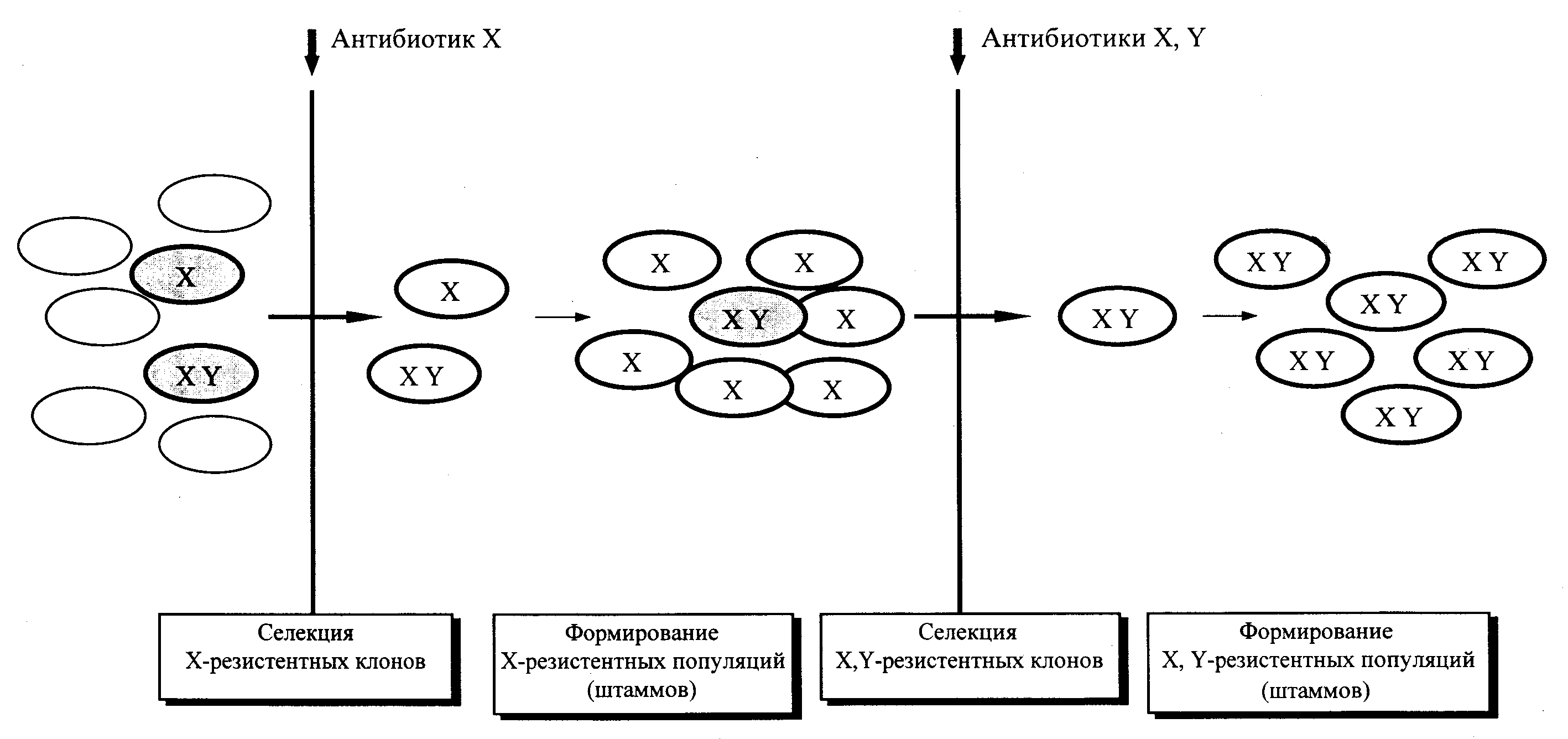
Бета-лактамные антибиотики. Яркой иллюстрацией к проблеме лекарственной устойчивости бактерий и борьбы с ней является история бета-лактамных антибиотиков — пенициллинов и цефалоспоринов. Эта борьба проходила и продолжается в двух направлениях — химическая модификация природных антибиотиков (получение более активных и устойчивых производных) и нейтрализация бета-лактамаз — самого важного фактора приобретенной бета-лактамной резистентности бактерий.
Природные пенициллины не действуют на большинство грамотрицательных бактерий и чувствительны к бета-лактамазам. Путем замены в пенициллановой кислоте бензильного кольца на другие радикалы были получены пенициллины новых поколений, отличающиеся друг от друга по антибактериальным спектрам, устойчивости к пенициллиназам и фармакологическим свойствам. В частности, удалось получить препараты, проникающие в грамотрицательные бактерии, значительно расширив спектр антибактериальной активности природного пенициллина. К таким антибиотикам относятся аминопенициллины, карбоксипенициллины и позже полученные уреидопенициллины (см. таблицу). Это производные ампициллина, в которых присутствие боковой цепи, связанной с аминогруппой на альфа-углероде, обеспечивает повышенную способность соединяться с пенициллинсвязывающими белками (транспептидазами) и ускоренное проникновение в грамотрицательные бактерии через поры в наружной мембране. Синтезирована и группа пенициллиназорезистентных пенициллинов, которые активны против бактерий, продуцирующих бета-лактамазы, прежде всего против пенициллиназопозитивных стафилококков.
Показательна и эволюция цефалоспоринов. Их изучение началось в 1945 г. после открытия антибактериальной активности секреторных продуктов гриба Cephalosporium, а клиническое применение началось с 1965 г. По сравнению с пенициллином, родоначальник современных цефалоспоринов более устойчив к инактивирующим ферментам, но менее активен. Добавление новых боковых цепей позволило получить цефалоспорины трех поколений. В цефалоспоринах 3-го поколения усилен ряд преимуществ, заложенных в препаратах 2-го поколения: они активны против синегнойной палочки и палочки инфлюэнцы, обладают повышенной устойчивостью к бета-лактамазам и хорошо проникают в центральную нервную систему. Последнее обстоятельство сделало их полезными при лечении грамотрицательного менингита. Однако не следует думать, что очередное поколение автоматически обесценивает предыдущие. Например, цефалоспорины 1-го поколения эффективны против грамположительных бактерий, тогда как препараты 2-го и 3-го поколений нацелены преимущественно на грамотрицательные бактерии. Поэтому иногда говорят не о поколениях, а о группах цефалоспоринов, отмечая микробиологические и фармакодинамические особенности (достоинства и недостатки) каждой из них.
Еще раз повторим, что вторичная устойчивость бактерий против пенициллинов и цефалоспоринов связана с плазмидозависимой (гораздо реже с хромосомной) продукцией бета-лактамаз — ферментов, которые разрушают активный центр бета-лактамных антибиотиков (рис. 12).

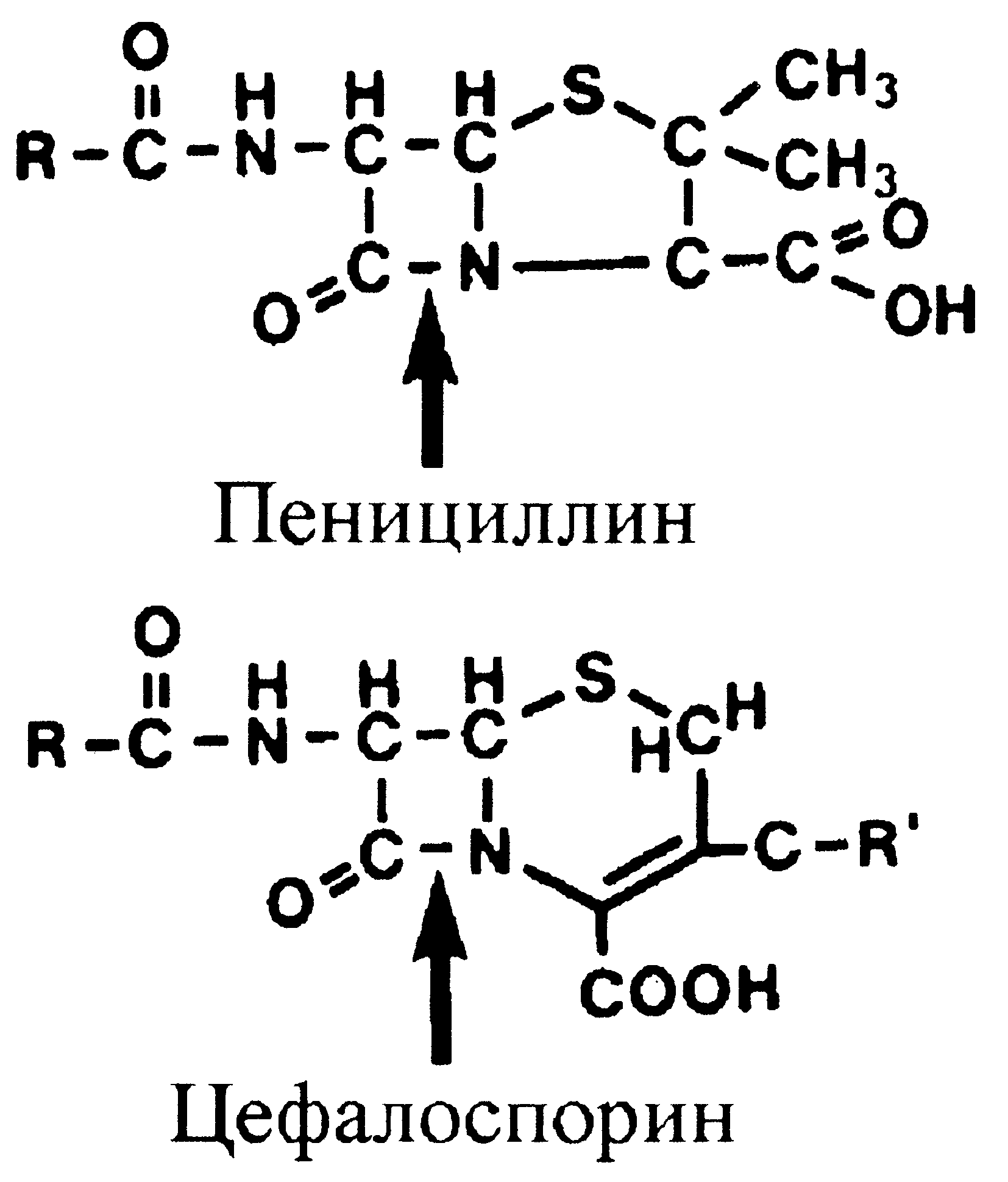
Известно более 100 бета-лактамаз, но далеко не все из них причастны к клинически значимой резистентности бактерий. Различают два вида бета-лактамаз — пенициллиназы и цефалоспориназы, что довольно условно, так как и те и другие атакуют обе группы антибиотиков, хотя и с разной эффективностью. Грамположительные бактерии (например, стафилококк) обычно продуцируют внеклеточные бета-лактамазы, которые разрушают препараты до контакта с бактериями. Они относятся к категории индуцибельных ферментов, причем в качестве индуктора нередко выступают сами антибиотики. В таких случаях повышение дозировки не усиливает антибактериального эффекта, так как ведет к гиперпродукции инактивирующего фермента. У грамотрицательных бактерий бета-лактамазы концентрируются в периплазме или связаны с плазматической мембраной. Они часто конститутивны, т.е. продуцируются на постоянном уровне, который не меняется под влиянием антибиотика. Поэтому повышение дозировки иногда помогает преодолеть резистентность. Можно, например, вспомнить о лечении гонореи: поначалу гонококк демонстрировал поразительную чувствительность к бензилпенициллину, но на протяжении последних 30 лет его дозировку приходилось постоянно увеличивать.
Печально известным примером быстрой эволюции резистентности к природным пенициллинам является стафилококк. Без проверки на чувствительность любой свежеизолированный штамм золотистого стафилококка сегодня рекомендуется считать пенициллиноустойчивым, чем фактически признается видовой ранг этого признака. Но и для других бактерий, которые недавно считались абсолютно чувствительными к бета-лактамам, появилось немало исключений. Штаммы, резистентные к пенициллинам, встречаются среди всех грамположительных и грамотрицательных бактерий, хотя пример катастрофически быстрого развития бета-лактамазной устойчивости уникален для стафилококка. Возможно, это связано с тем, что у многих бактерий резистентность возникла на хромосомной основе, и лишь позднее стали доминировать штаммы с мобильными r-генами. Кроме того, бета-лактамные антибиотики различаются по аффинности к бета-лактамазам. Некоторые из них (пенициллиназоустойчивые пенициллины, цефалоспорины 3-го поколения, имипенем) гидролизуются немногими бета-лактамазами (их называют бета-лактамазоустойчивыми), тогда как другие (например, ампициллин) гораздо чувствительнее. Есть антибиотики, которые устойчивы против грамотрицательных бета-лактамаз, но разрушаются грамположительными бактериями (например, темоциллин).
Для подавления активности бета-лактамаз в состав препаратов предложено включать их ингибиторы — клавулановую кислоту и сульфоны пенициллановой кислоты (сульбактам, YTR-830 и др.). Они относятся к семейству бета-лактамов, но обладают слабой антибиотической активностью. Вместе с тем, располагая бета-лактамным кольцом, они великолепно реагируют с бета-лактамазами и, перехватывая их, предотвращают разрушение «настоящих» антибиотиков. Фермент и ингибитор могут вступать во временную связь с образованием непрочного комплекса, но чаще происходит необратимая инактивация фермента. Спектр ингибируемых бета-лактамаз весьма широк, включая наиболее распространенные грамположительные и грамотрицательные бета-лактамазы. Может показаться странным, что, располагая возможностью получения устойчивых мономолекулярных антибиотиков, идут по пути создания сложных смесей. Но дело в том, что структурные изменения, благодаря которым достигается бета-лактамазная резистентность, иногда негативно влияют на антибактериальные и фармакологические свойства препарата (например, активность пенициллиназоустойчивых пенициллинов в 10—30 ниже природного пенициллина). Сочетание с ингибиторами позволяет избежать этого, используя достоинства классических бета-лактамов.
Гликопептидные антибиотики. В редких случаях устойчивость к бета-лактамам связана со снижением проницаемости бактериальных клеток или мутациями по пенициллинсвязывающим белкам. Эти механизмы давно вызывают серьезную озабоченность в связи с так называемыми метициллинрезистентными штаммами золотистого стафилококка (см. «Стафилококк»). Для борьбы с ними приходится применять другие антибиотики, прежде всего ванкомицин. К сожалению, сегодня известны бактерии (прежде всего энтерококки и золотистые стафилококки), которые приобрели резистентность к гликопептидным антибиотикам, в частности к ванкомицину. Она связана с заменой d-Ala-d-Ala в мурамилпептиде пептидогликана на d-Ala-d-лактат. Он не взаимодействует с ванкомицином, препятствуя его антибактериальному эффекту. Это достигается благодаря совместному действию нескольких ферментов — VanA/VanB, VanH и VanX. Заключительную операцию выполняет VanX, который разрушает остаточные связи d-Ala-d-Ala, оставляя интактными группы d-Ala-d-лактат, образующиеся под действием VanA/VanB и VanH.
Тетрациклины. Устойчивые штаммы не аккумулируют тетрациклин. Парадоксально, но это связано не со снижением проницаемости клеточной стенки, а с ускоренной экскрецией антибиотика. Этот механизм обусловлен плазмидозависимой перестройкой цитоплазматических белков и работает почти у всех бактерий — грамположительных и грамотрицательных, аэробных и анаэробных. Бактероиды обезвреживают тетрациклины и более традиционным способом — путем химических модификаций.
Хлорамфеникол (левомицетин). Устойчивость к хлорамфениколу приобрели многие бактерии. Печальным примером, который заставил обратить на это внимание, явились эпидемические вспышки тяжелой дизентерии и брюшного тифа в Центральной Америке и Мексике в конце 1960-х — начале 1970-х гг. Хлорамфеникол (он считался тогда лучшим средством против шигелл и сальмонелл) получали все больные, но вопреки ожиданиям лечение было неэффективным и многие погибли. Это говорит о том, какой угрозой может стать неожиданное появление резистентных штаммов среди эпидемичных видов бактерий. Устойчивость объясняется ацетилированием гидроксильных групп хлорамфеникола плазмидозависимым ферментом — специфической ацетилтрансферазой. Ацетилированный хлорамфеникол не активен, так как не связывается с рибосомами.
Макролиды и линкозамиды. Химической модификации (метилированию) подвергается внутриклеточная мишень — 23S-рибосомальная РНК (она входит в состав 50S-субъединицы рибосом), причем для приобретения устойчивости достаточно метилирования всего двух адениннуклеотидов. Метилаза закодирована в индуцибельной R-плазмиде, и ее синтез быстро возрастает при добавлении антибиотиков, особенно эритромицина. Слабыми индукторами метилазы являются линкозамиды, прежде всего клиндамицин. Поэтому штаммы, устойчивые к эритромицину, могут имитировать псевдочувствительность к клиндамицину in vitro, хотя в организме они резистентны.
Аминогликозиды. В приобретении устойчивости к аминогликозидам участвуют разные механизмы. Наиболее важным является изменение структуры антибиотиков — фосфорилирование, аденилирование или ацетилирование гидроксильных и аминогрупп. Гены для этих ферментов часто закодированы в плазмидах (транспозонах) и передаются между видами. Ослабление активности объясняется тем, что модифицированные антибиотики плохо проникают в бактериальные клетки. Новое поколение полусинтетических аминогликозидов (см. таблицу) имеет специальные «хвосты», которые повышают устойчивость против инактивирующих ферментов. Резистентность грамотрицательных бактерий дополнительно усиливается благодаря снижению проницаемости клеточной стенки и ослаблению трансмембранного транспорта аминогликозидов. Наконец, возможны мутации по рибосомным белкам, от которых зависит связывание аминогликозидов с рибосомами. Бактерии довольно широко используют этот способ для защиты от стрептомицина и редко — от других аминогликозидов. Замена единственной аминокислоты в протеине р10, входящем в состав 30S-субъединицы, блокирует связывание стрептомицина, снимая его антибактериальный эффект.
* * * Приобретение резистентности следует считать биологической закономерностью, которая, хотя и в разной степени, справедлива для всех бактерий и всех антибиотиков. Это часть общебиологической проблемы, связанной с адаптацией живых организмов к экстремальным условиям внешней среды. К антибиотикам адаптируются не только бактерии, но и остальные микроорганизмы — от эукариотических форм (простейшие, грибы) до вирусов. Прокариоты располагают скрытым потенциалом высокомобильных генов резистентности, которые оживают под влиянием антимикробных агентов и получают быстрое распространение на внутривидовом и межвидовом уровнях. Одним из резервуаров r-генов служит нормальная микрофлора, изобилующая многочисленными видами бактерий. Здесь складываются благоприятные условия для обмена транспозонами и плазмидами, тем более, что эндогенные бактерии часто попадают под косвенную атаку антибактериальных агентов и вынуждены выживать в их присутствии, напрягая генетические резервы адаптации. Шок от первых столкновений с приобретенной резистентностью бактерий прошел. Раскрыты ее основные механизмы, известны приемы, которыми можно бороться с этим явлением. Ясно и то, что борьба будет вечной, как вечны жизнь и стремление к ней всего живого.
Вирусы гепатита
- Характеристика известных гепатотропных вирусов.
- Острые и хронические гепатиты.
- Степени риска.
- Эпидемиология и профилактика.
- Специфическая диагностика.
- Вирусы, претендовавшие/претендующие на избирательную гепатотропность.
Понятие «гепатит» означает повреждение печеночной паренхимы, т.е. гепатоцитов. Это наблюдается при многих инфекционных заболеваниях, но есть инфекционные агенты, для которых поражение печени — единственный, или по крайней мере главный патогенетический механизм, определяющий всю или почти всю клиническую картину заболевания. Так ведет себя группа вирусов, которые, обладая избирательной гепатотропностью, инфицируют преимущественно гепатоциты. С ними связано большинство инфекционных поражений печени: от стертых до фульминантных форм, от спорадических случаев до эпидемий. Нередко инфекция продолжается после завершения острого процесса или стабилизируется без его проявлений. Это характерно для вирусов с парентеральным механизмом передачи, которые длительно (нередко пожизненно) персистируют в гепатоцитах, поддерживая хронический процесс и угрожая тяжелейшими осложнениями.
Социально-экономические потери от вирусных гепатитов огромны. Они обусловлены глобальным распространением вирусов и их эпидемиологическим коварством, сопряженным с широкой прослойкой бессимптомных и малосимптомных инфекций. Несомненно, это одна их самых распространенных инфекционных патологий и одна из важнейших проблем здравоохранения.
Вирусные гепатиты представляют группу нозологически самостоятельных заболеваний печени. Их разграничение, инициированное эпидемиологами, получило реальную основу после открытия дискетных возбудителей и разработки лабораторных тестов для их обнаружения в инфицированном материале и окружающей среде. Внедрение в практику серологических тестов для диагностики вирусных гепатитов С и Е значительно сократило число этиологически не расшифрованных случаев острой и хронической патологии печени. Вместе с тем до сих пор 10—20% гепатитов, инфекционная природа которых очевидна, остаются этиологически не распознанными. Допускается существование новых вирусов, вызывающих гепатиты с энтеральным и парентеральным механизмами заражения. Формула ни А—В-гепатиты трансформирована в новое понятие — ни А—Е-гепатиты, но скорее всего и оно не будет последним.
Сегодня просматриваются хорошие перспективы для успешного решения спектра проблем, связанных с вирусными гепатитами. С этой целью следует разумно использовать блестящие достижения современной вирусологии, иммунологии и молекулярной биологии, благодаря которым созданы уникальные возможности для диагностики и предупреждения вирусных инфекций.
Вирус гепатита А
Вирус гепатита А (HAV) относится к семейству пикорнавирусов, где числится как единственный представитель рода Hepatovirus. С рядом исключений (своеобразная структура РНК, высокая устойчивость во внешней среде, необычные ростовые свойства) он повторяет главные генетические и фенотипические признаки энтеровирусов, в частности полиовируса — возбудителя полиомиелита. Его вирион представляет собой голый (безоболочечный) икосаэдральный нуклеокапсид диаметром 27—30 нм. Он включает геномную однонитчатую РНК (около 7500 нуклеотидных оснований, т.е. 7,5 кбайт) и пять структурных белков, четыре из которых (VP1—VP4) образуют капсид, выполняя прикрепительную и адгезивную функции (избирательное связывание с гепатоцитами). Один из вирионных белков (VPg) связан с РНК, участвуя в ее внутриклеточных превращениях. Капсидные белки объединяются в понятие «антиген вируса гепатита А» (HAAg). HAV включает 7 генотипов, имеющих до 85% РНК-гомологии; большинство инфекций у человека вызывают вирусы I и III генотипов. Генотипирование может иметь эпидемиологическое, но не клиническое значение, так как все штаммы HAV имеют единый серотип HAAg, который детерминирует образование общих вируснейтрализующих антител. После проникновения в клетку РНК вириона подвергается трансляции (т.е. является плюс-нитевой) с образованием полипротеина (пропептида), из которого при участии клеточных протеаз вычленяются вирусспецифические ферменты — РНК-полимераза и протеаза. РНК-полимераза обеспечивает репликацию геномной молекулы, а вирусная протеаза завершает расщепление первичного белкового продукта с образованием структурных полипептидов (рис. 1).
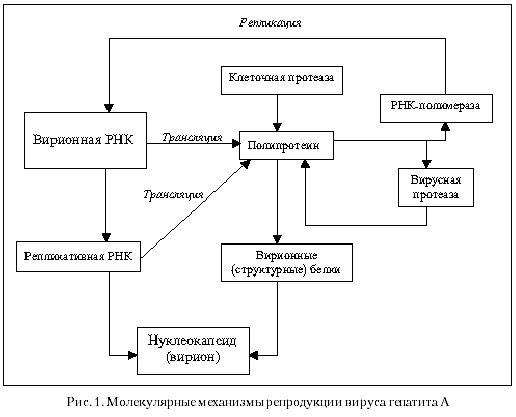

HAV — единственный из вирусов гепатита, который удается культивировать in vitro. Однако в отличие от других энтеровирусов он не вызывает деструкции зараженных клеток, а имеет тенденцию к персистенции. Это не соответствует его взаимоотношениям с гепатоцитами in vivo, которые остро погибают от продуктивной вирусной инфекции. В связи с этим главной моделью для изучения HAV служит заражение обезьян (шимпанзе и мармозетов).
Гепатит А — кишечная инфекция с фекально-оральным механизмом передачи. Возбудитель передается через зараженную воду, пищу и контактно-бытовым путем. Этому способствует устойчивость вируса во внешней среде и высокая восприимчивость к нему человека. Вирус имеет глобальное распространение: с ним контактирует 70—90% населения планеты (по отдельным регионам показатель колеблется от 13 до 99%). Этим объясняется тот факт, что по заболеваемости гепатит А лидирует среди всех вирусных гепатитов. В регионах с низким санитарным уровнем большинство людей инфицируется в раннем детстве. В этом случае заражение обычно протекает бессимптомно, оставляя пожизненный иммунитет. В социально благополучных группах первичному инфицированию чаще подвергаются взрослые (обычно в возрасте 15—25 лет), что нередко ведет к манифестным формам. Тем не менее гепатит А считается преимущественно детской инфекцией, в структуре которой большинство составляют школьники начальных классов.
Для гепатита А характерны эпидемиологические признаки любой кишечной инфекции. Заболеваемость может ограничиться спорадическими случаями, но всегда остается угроза вспышек и даже эпидемий. Подъем заболеваемости начинается в июле-августе, достигает максимума в октябре-ноябре. Эффективность противоэпидемических мероприятий снижается из-за высокого числа безжелтушных (стертых) форм, которые, по некоторым данным, составляют 90—99%. Кроме того, наиболее интенсивное выделение вируса с фекалиями происходит в конце инкубационного периода, т.е. до появления клинических симптомов.
В отличие от других энтеровирусов (ранее он числился как энтеровирус 72) HAV не размножается (или почти не размножается) в эпителиоцитах слизистой оболочки кишечника, а сразу транспортируется через воротную вену в печень. Это означает, что кишечная локализация HAV фактически отсутствует. Пищеварительный тракт служит главным образом местом внедрения вируса в организм и резервуаром, куда он выделяется после репликации в печени. Инкубационный период колеблется от 10 до 50 дней, но обычно не превышает месяца. Клиническим симптомам предшествует период нецитопатической (предморфологической) репликации вируса в гепатоцитах, который продолжается 7—10 дней. Ему сопутствуют накопление сывороточных маркеров повреждения гепатоцитов (в частности, трансаминаз), вирусемия и выход вируса с желчью в кишечник. Короткая фаза вирусемии объясняет редчайшие случаи парентерального заражения гепатитом А.
За периодом скрытой репликации следует поражение гепатоцитов с развитием клиники острого гепатита. С этого момента больной практически не заразен для окружающих, так как вирусемия, а главное выделение вируса с фекалиями резко идут на убыль. Этот парадокс отражает иммунологический контроль над HAV-инфекцией, активность которой быстро падает с появлением симптомов клинического кризиса. Высокий уровень инфекционного вируса в печени до развития гепатита, а также отсутствие цитопатических изменений в зараженных клеточных культурах позволяют думать об иммунологически зависимом поражении печеночной паренхимы. В пользу этого говорят и наблюдения о присутствии в крови больных гепатитом А цитотоксических Т-лимфоцитов, агрессивно настроенных против HAV-инфицированных клеток.
Объем повреждения печени почти всегда ограничен, не выходя за рамки фокального некроза. Антитела появляются уже в инкубационном периоде, блокируя распространение вируса. Клиническое выздоровление знаменует полное освобождение от HAV. Персистенция вируса и ее осложнения (хронический гепатит, цирроз, гепатома) не встречаются. Фульминантный гепатит (массивный некроз гепатоцитов) наблюдается исключительно редко.
Для иммунопрофилактики гепатита А предложена вакцина, инактивированная формалином, которая хорошо зарекомендовала себя на практике. Однако для получения длительного эффекта требуются повторные введения. Этим объясняются попытки разработать и внедрить живую аттенуированную вакцину для парентерального или перорального применения.
Полезным средством специфического воздействия на эпидемический процесс является обычный иммуноглобулин человека. Вместе с тем не следует забывать, что, подобно любой кишечной инфекции, гепатит А вполне управляем в системе общего противоэпидемического надзора и комплекса мероприятий, нацеленных на повышение санитарной культуры населения. Абсолютным подтверждением клинического диагноза служит обнаружение вируса и/или его антигенов (HAAg) в экстрактах фекалий. При этом следует помнить об эпидемиологическом парадоксе гепатита А: фаза интенсивного (диагностически значимого) выделения возбудителя совпадает с окончанием инкубационного периода и прекращается с нарастанием клинических симптомов. Поэтому отсутствие вируса в испражнениях (об этом судят по отрицательным данным электронной микроскопии, полимеразной цепной реакции и выявления HAAg) не исключает заболевания.
Надежным методом служит определение анти-HAV IgM-антител в сыворотке. Они появляются уже в инкубационном периоде, что позволяет не только диагностировать гепатит А, но и выявлять малосимптомные случаи свежей инфекции. Обнаружение анти-HAV класса IgG является индикатором перенесенной инфекции и устойчивости к повторному заражению. Нарастание титра IgG анти-HAV говорит об острой инфекции, но в связи с поздним поступлением больных в стационар такую динамику уловить трудно.
Таблица 1
Маркеры гепатита А
| Период болезни | Вирус/HAAg в фекалиях | IgM анти-НAAg | IgG анти-НААg |
| Инкубационный период | + | + | – |
| Клинически выраженный гепатит | – | + | + |
| Реконвалесценция | – | +/– | + |
| Постинфекционный период | – | – | + |
Характеристика специфических маркеров HAV-инфекции в проекции на инфекционный процесс представлена в таблице 1.
Вирус гепатита В
Вирус гепатита В (HBV) представляет единственный вид рода Orthohepadnavirus cемейства гепатотропных ДНК-содержащих вирусов, Hepadnaviridae, которое объединяет несколько сходных вирусов млекопитающих и птиц (вирусы гепатита сурков, земляных и древесных белок, пекинских уток и цапель). Они используются в модельных опытах по изучению биологии гепаднавирусов, так как сам HBV не удается культивировать in vitro за исключением обычно инфицированных им клеток первичной гепатомы.
Вирус гепатита В — один из мельчайших оболочечных вирусов человека. Его вирион (частица Дейна) имеет сферическую форму диаметром 40—45 нм. Под электронным микроскопом в вирионах отчетливо видны сердцевина (кор, т.е. нуклеокапсид) и наружная липопротеиновая оболочка (суперкапсид) (рис. 2).
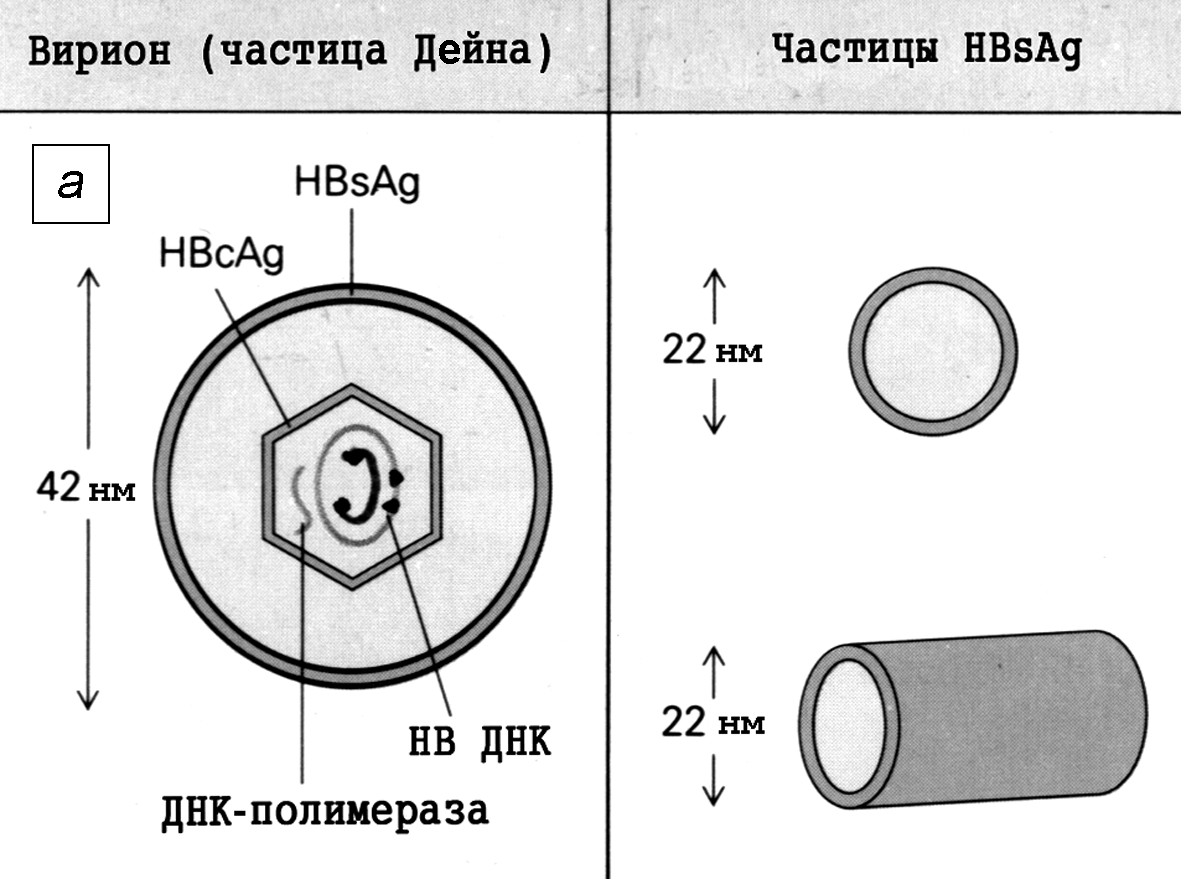

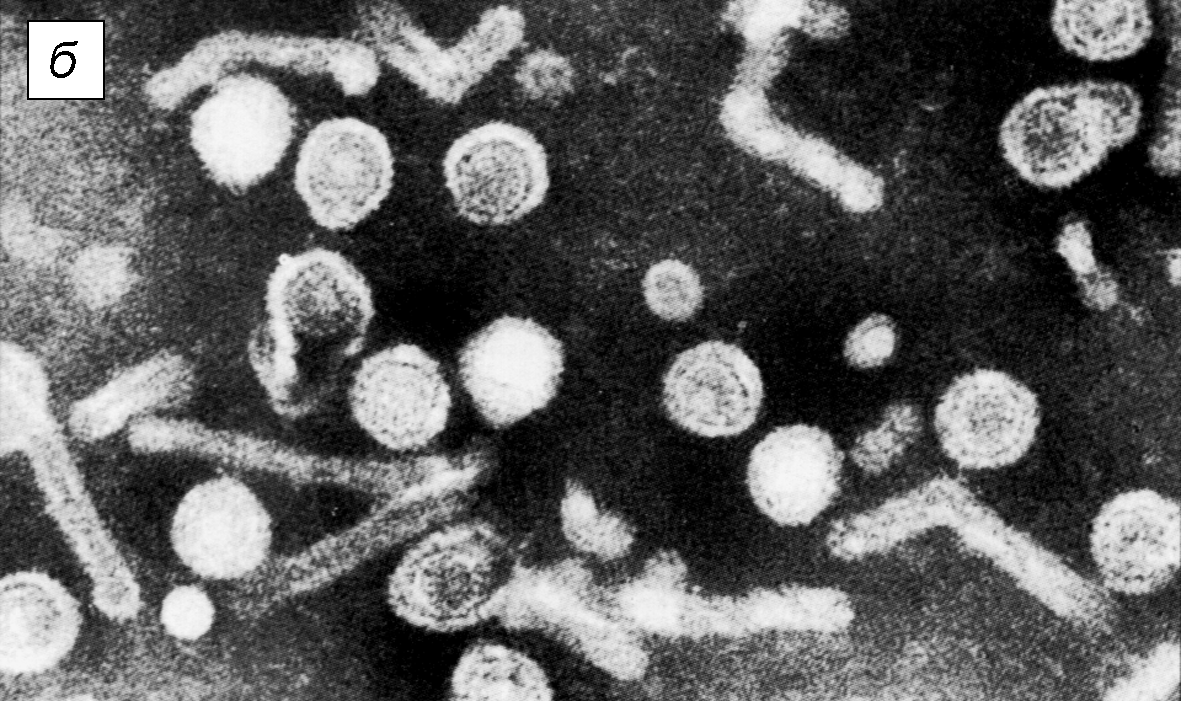

Рис. 2. Вирион вируса гепатита В (частица Дейна) и HВsAg-частицы:
а — принцип строения; б — электронограмма крови больного
Вирусные белки. Вирус обладает пятью белковыми антигенами: HBsAg, HbcAg, HBeAg, HBxAg и HBpol.
HBsAg (устар. австралийский антиген) — поверхностный антиген (s от англ. surface — поверхность). Обеспечивает избирательное прикрепление вируса к мембране гепатоцитов, возможно за счет связывания с полимеризованным сывороточным альбумином.
Понятие «HBsAg» объединяет три оболочечных гликозилированных белка — S, M и L. Они являются производными s-гена и прилежащих к нему коротких ДНК-фрагментов, pre-s1 и pre-s2. Их экспрессия дает три продукта — HBsAg-S (производное s-гена; англ. short — короткий), HВsAg-M (продукт s- и pre-s1; англ. middle — средний) и НВsAg-L (продукт s-, pre-s1 и pre-s2; англ. long — длинный, большой). В вирионе 70% приходится на HВsAg-S — главный оболочечный антиген. НВsAg-M и HBsAg-L cоставляют примерно 30% поверхностного белка. HBsAg-L принимает участие в стыковке суперкапсида с кором и, возможно, в рецепции вириона клетками печени. Функции HBsAg-M не известны.
HBsAg cодержит протективные В-эпитопы и индуцирует синтез антител, блокирующих прикрепление вируса к рецепторам гепатоцитов. Синтезируется в избытке (особенно при интегративной инфекции — см. ниже) и в значительных количествах секретируется в кровь, где определяется не только в структуре вирионов, но и в виде свободных агрегатов (сферические частицы, филаменты), состоящих из S- и L-белков. Их количество во много раз превосходит число полноценных вирионов, достигая 1012 в 1 мкл сыворотки (см. рис. 2).
По HВsAg-эпитопным особенностям дифференцируют четыре основных HBV-серовара, которые используются как эпидмаркеры. По меньшей мере один из эпитопов (a) является общим для всех штаммов вируса, что обеспечивает возможность универсальной диагностики, основанной на выявлении HВsAg в сыворотке, а также применение стандартной HВsAg-вакцины для профилактики гепатита В.
HВcAg, или кор-антиген — главный белок HBV-нуклеокапсида, или сердцевины (англ. core — сердцевина). Кодируется c-геном, к которому прилежит небольшой, но функционально значимый фрагмент ДНК — пре-c. От того, с какого участка пре-c начинается транскрипция мРНК, зависит качество и судьба НВсAg. Один из его вариантов входит в состав вирионного нуклеокапсида, другой (гидрофобный) включается в клеточные мембраны и служит объектом для распознания и атаки Т-лимфоцитами. В кровь не секретируется и в свободном состоянии не определяется.
HBeAg — важный для диагностики (и, по-видимому, для репликации вируса) продукт c-гена (англ. envelope — оболочка). Образуется в результате протеолиза (возможно, аутопротеолиза) кор-антигена, встроенного в клеточные мембраны. Отличается от последнего по В-эпитопам (т.е. к каждому из них образуются собственные антитела), но содержит общие с ним Т-эпитопы. Не входит в состав вириона, а выделяется в кровь, где его содержание коррелирует с активностью вирусной репликации.
HBхAg — неструктурный белок. Определяется в сыворотке и печени при высоком уровне вирусной репликации. Выполняет функции трансактиватора вирусных и клеточных генов, способствуя развитию патологического процесса в печени. Его производные определяются в мембране гепатоцитов почти у половины больных хроническим гепатитом, являясь объектом для цитотоксических Т-лимфоцитов. Полагают, что наряду с инсерционным мутагенезом (результат внедрения/инсерции вирусного генома в хромосомы клетки) HBxAg участвует в индукции злокачественного перерождения HBV-инфицированных гепатоцитов, активируя клеточные онкогены и блокируя антионкогенные (апоптозиндуцирующие) белки, в частности р53.
HBVpol (полимераза) — полифункциональный фермент, входящий в состав HBV-сердцевины. Продукт самого крупного, р-гена, перекрывающего s- и c-локусы. Обладает активностью ДНК-полимеразы, обратной транскриптазы и РНК-азы (см. ниже). Играет ключевую роль на всех этапах репликации вируса.
Геном. Геном гепаднавирусов устроен своеобразно. Он состоит из двух ковалентно незамкнутых нитей ДНК, одна из которых (плюс-нить) не достроена, т.е. короче другой (минус-нити) на 20—30%. Ликвидация этого дефекта является обязательным этапом репликации вируса и обеспечивается вирионной ДНК-полимеразой после проникновения вируса в ядро гепатоцитов. Каждому из главных вирусных белков соответствует открытая рамка считывания (фактически ген), но небольшой размер генома (3,2 кбайт, т.е. меньше, чем у любого из известных вирусов с двухспиральной ДНК) не позволяет синтезировать даже столь скромный набор антигенов без дополнительных ухищрений, нацеленных на увеличение генетической емкости ДНК. Это достигается путем транскрипции с перекрывающихся областей ДНК (перехлест генов) и сдвига рамок трансляции мРНК-транскриптов. Кроме того, синтезированные полипептиды подвергаются посттрансляционным модификациям (ограниченному протеолизу, гликозилированию), меняющим их структуру и функции.
Репликативная инфекция. Репликация HBV происходит необычно. Это единственный из ДНК-вирусов человека, который реплицируется с подключением механизма обратной транскрипции по схеме ДНК—РНК—ДНК (рис. 3). После достраивания в ядре дефектной ДНК-нити она используется клеточной РНК-полимеразой в качестве матрицы для синтеза полной РНК-копии — прегенома (pgRNA). Одновременно образуются и более короткие РНК, которые подвергаются трансляции, обеспечивая синтез вирусных белков. pgRNA транспортируется в цитоплазму и после объединения с капсидом переписывается в ДНК, т.е. подвергается обратной транскрипции. РНК разрушается, и на свободных участках ДНК строится вторая (дефектная) ДНК-нить.

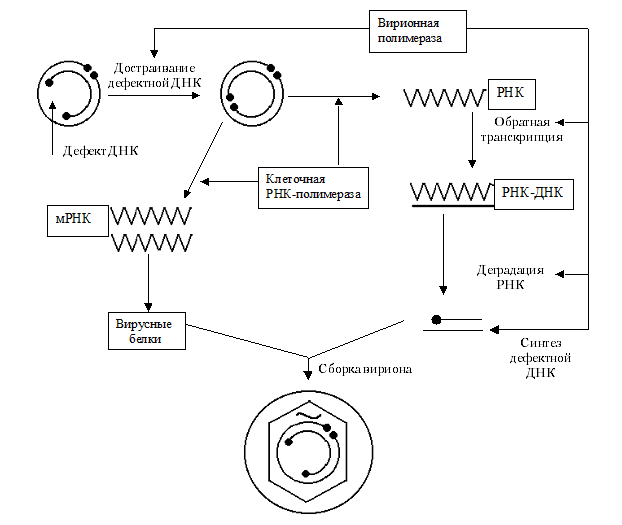
Все перечисленные этапы (достраивание дефектной ДНК-нити, обратная транскрипция, разрушение pgRNA, синтез дефектной ДНК-нити) обеспечиваются общим ферментом — вирионной полимеразой. Полифункциональность снижает точность ферментативных реакций. Весомость ошибок возрастает, если они (как в данном случае) допускаются при репликации. С этим, в частности, может быть связана высокая мутационная изменчивость HBV, которая накладывает серьезный отпечаток на взаимоотношения с хозяином. Механизм обратной транскрипции сближает гепаднавирусы с ретровирусами. По этой причине их даже предложено называть параретровирусами.
Процесс репликации заканчивается сборкой суперкапсида. Кор одевается наружной оболочкой, используя для этого внутриклеточные мембраны аппарата Гольджи с встроенными в них HВsAg (S, L и М), и высвобождается из клетки.
Интегративная инфекция. Кроме репликативной инфекции, которая ведет к воспроизведению вирионов, вирус гепатита В имеет в арсенале еще одну стратегию — он охотно встраивает свою ДНК в геном зараженной клетки, используя ее как прикрытие. Этот механизм, хорошо известный в вирусологии, носит название интегративной вирогении. Не обеспечивая репродукции (хотя такая возможность остается), он способствует длительному сохранению (персистенции) вируса в инфицированных клетках, что поддерживает его патогенетический потенциал. Экспрессия вирусных генов блокируется; исключение составляют s- и, по-видимому, x-гены. Транскрипция s-гена даже усиливается, если он попадает под контроль клеточных генов-промоторов. Образующийся в избытке HBsAg не накапливается в гепатоцитах, а в больших количествах выделяется в кровь. При хроническом гепатите репликативная и интегративная инфекции развиваются параллельно, но со временем верх берет интегративный компонент, который существенно влияет на развитие цирроза печени и первичной гепатомы.
Патология. Вирусом гепатита В заражено около 5% населения Земного шара; в отдельных регионах этот показатель колеблется от 1 до 20% и более. Большинство из инфицированных не имеют в анамнезе острого гепатита, а становятся носителями после субклинической инфекции. Способность персистировать в гепатоцитах — характернейший признак HBV. С ним связана патогенетическая стратегия вируса и его длительное выживание в условиях явного или скрытого патологического процесса.
Взаимоотношения HBV с человеком отражены на рис. 4.

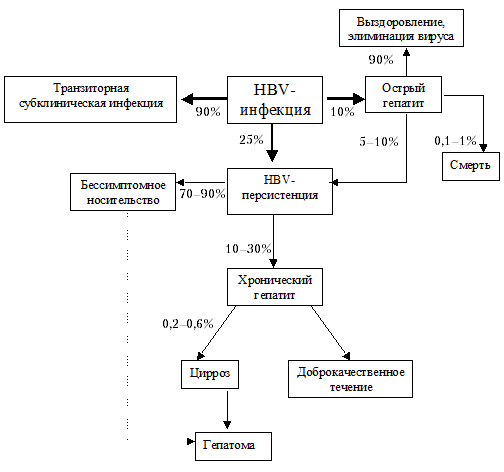
Контакт с вирусом обычно ведет к бессимптомной инфекции. Острый гепатит развивается после длительной инкубации (6 нед — 6 мес) примерно у 10% инфицированных и в большинстве случаев заканчивается выздоровлением. Но клиническая реконвалесценция не всегда сочетается с вирусологическим выздоровлением, т.е. с удалением вируса из организма: у 10% зараженных (включая лиц с бессимптомной инфекцией и больных, перенесших острый гепатит) формируется хроническая (персистентная) инфекция. О ней принято говорить, если HBsAg сохраняется в сыворотке крови более 6 мес.
Застрявший в гепатоцитах вирус может спонтанно элиминироваться, но часто этого не происходит, и он надолго (нередко навсегда) остается в печени, подвергаясь репликации и/или интегративной вирогении. Обычно присутствие вируса клинически себя не проявляет, но у 10—30% носителей формируется хронический гепатит с более или менее агрессивным течением. При доброкачественных формах допускается возможность самоизлечения или, по крайней мере, длительных ремиссий, но эволюция процесса может быть и крайне неблагоприятной, завершаясь необратимым циррозом печени и малигнизацией гепатоцитов с развитием первичного рака печени — гепатомы. Вероятность ее возникновения у HBsAg-носителей в 200—300 раз выше, чем у лиц, свободных от вируса, причем риск особенно велик при заражении в перинатальном периоде или в раннем детстве.
За внешними (клиническими, морфологическими, биохимическими) проявлениями хронической HBV-инфекции стоят сложные взаимоотношения между вирусом, зараженной клеткой и системой иммунологического надзора. Вирус пользуется разнообразными способами, чтобы поддержать персистенцию, включая мутационные изменения антигенов, вирогению, уклонение от иммунного ответа, нейтрализацию эффекторов иммунитета. Но несмотря на это многие гепатоциты, несущие вирус, распознаются системой иммунитета и подвергаются уничтожению через Т-зависимый цитолиз и/или апоптоз (прямая цитотоксичность у HBV выражена слабо). Этот позитивный (с точки зрения элиминации вируса) механизм служит одновременно патогенетической основой для формирования хронического гепатита, активность которого в целом отражает интенсивность внутрипеченочной репликации вируса. В связи с этим кроме клинико-морфологических признаков в классификации хронического HBV-гепатита учитываются вирусологические данные, прежде всего качество репликативного процесса в гепатоцитах. Об этом можно судить, исследуя биоптаты печени, но гораздо доступнее кровь, в которую попадают зрелые вирионы (вирусемия) и их антигены (антигенемия). Практически важным является разграничение HBeAg-позитивного и HBeAg-негативного хронических гепатитов, которые соответствуют репликативной и интегративной формам персистентной HBV-инфекции. Хронический гепатит с HBeAg-антигенемией протекает обычно тяжелее, с большей склонностью к цирротическим изменениям печени. Обнаружение HBeAg в крови является показанием к противовирусной терапии (интерферонотерапия, аналоги нуклеозидов).
Эпидемиология. Эпидемиологию гепатита В определяют следующие факторы:
- широкое распространение вируса;
- в естественных условиях вирус опасен только для человека (из экспериментальных животных к нему чувствительны шимпанзе);
- высокий процент здоровых носителей, которые длительно (часто пожизненно) остаются нераспознанными;
- высокая чувствительность человека к возбудителю, допускающая возможность заражения ничтожными дозировками вируса (например, при попадании в организм 0,0001 мл HBsAg-позитивной плазмы);
- высокая концентрация инфекционного вируса в крови (при заражении шимпанзе контагиозность сыворотки гепатитных больных сохраняется в разведении до 10-7—10–8);
- присутствие эпидемически значимой концентрации вируса в сперме, грудном молоке и, возможно, слюне;
- высокая устойчивость вируса во внешней среде и к стерилизующим процедурам;
- множество естественных и искусственных путей передачи возбудителя;
- отсутствие механизма биогенной трансмиссии (через кровососущих насекомых).
Из этого перечня следует, что гепатит В — антропонозная инфекция, при которой решающим механизмом передачи служит парентеральное инфицирование, т.е. проникновение возбудителя в кровяное русло через естественные или искусственные повреждения кожи и слизистых оболочек. Главным источником инфекции является кровь носителей вируса. Это оправдывает характеристику гепатита В как кровяной инфекции или сывороточного гепатита. Вероятность заражения при контакте с другими биологическими субстратами (прежде всего спермой) тоже реальна, хотя по значимости они уступают крови и ее дериватам. Гепатит В — глобальная инфекция, но ее распространение значительно колеблется в отдельных регионах. Заболеваемость (не считая редких исключений) носит спорадический характер и не имеет сезонного характера.
Реализация парентерального механизма передачи HBV достигается разными способами:
- медицинские процедуры (гемотрансфузия, гемодиализ, инъекции, инструментальная диагностика, стоматологические операции и пр.);
- бытовое инфицирование (татуировка, маникюр, педикюр, бритье, пользование общими туалетными принадлежностями — зубные щетки, бритвы, мочалки и пр.);
- половой путь (вирус из крови или спермы проникает через микротравмы слизистых оболочек при половых контактах);
- трансплацентарная передача и заражение в родовом канале;
- инфицирование в неонатальном периоде (через микротравмы при кормлении, грудное молоко, слюну).
Cпецифическая диагностика (выявление вирусных субкомпонентов). Одной из важнейших особенностей гепатита В является то, что вирус и его субкомпоненты в больших количествах выделяются в кровь. В крови зараженных людей почти всегда присутствуют зрелые HBV-вирионы (вирусемия) и его антигены (антигенемия). Это создает отличные возможности для диагностики, наблюдения за развитием инфекционного процесса, выявления соотношений между репликативной и интегративной инфекциями, определения степени эпидемиологической опасности реконвалесцентов и носителей HBV-инфекции.
HBV-вирионы (частицы Дейна) — абсолютный критерий вирусемии, который отражает репликацию вируса в гепатоцитах. Из-за сложности (необходима электронная микроскопия) метод не используется в практических лабораториях.
HBV-ДНК — абсолютный и высокочувствительный критерий репликативной инфекции и вирусемии. Являясь геномной молекулой, ДНК всегда ассоциирована с инфекционным вирусом. Ее определение основано на полимеразной цепной реакции.
мплифицированные (т.е. многократно прокопированные в системе со специфическими праймерами) ДНК-фрагменты идентифицируются относительно стандартных ДНК-проб, которые по последовательности нуклеотидов гомологичны искомым ДНК-фрагментам. Доступность метода ограничивается необходимостью сложного оборудования и дорогостоящих реактивов.
НВsAg — наиболее популярный и универсальный маркер HBV-инфекции. Синтезируется в избытке при репликативной инфекции, а после включения вирусной ДНК в состав клеточных хромосом может быть единственным продуктом вирусного генома. Этим определяется обязательное присутствие HBsAg в крови больных острой и хронической HBV-инфекцией в концентрациях, иногда сопоставимых с содержанием собственных белков плазмы. При остром гепатите обнаруживается в сыворотке за несколько недель до клинических симптомов (раньше частиц Дейна, вирусной ДНК и HBeAg) и исчезает почти одновременно с выздоровлением, но позже других компонентов вируса. Освобождение от HBsAg сочетается с появлением анти-HBs-антител. Нередко они выявляются с опозданием, спустя некоторое время после удаления из циркуляции HBsAg. Этот период, когда гепатит В не диагностируется при помощи тестов на HBsAg и анти-HBs, известен как HBV-окно. В таких случаях диагноз верифицируется по антителам (лучше IgM) к HBcAg, которые появляются в разгар заболевания. Негативные тесты на НВsAg при повторных анализах в сочетании с положительной пробой на анти-HBs служат гарантией вирусологического излечения от гепатита В, т.е. удаления вируса из организма. Сохранение HBsAg-позитивности более 6 мес считается признаком хронической инфекции. Так как стабилизация диагностических титров HBsAg в крови возможна без репликации вируса или при ее низкой активности, для количественной оценки репликативного процесса требуется анализ других маркеров, которые абсолютно или тесно коррелируют с вирусемией.
HВcAg — теоретически возможный маркер HBV-репликации и вирусемии. Для его выявления в сыворотке требуется разрушение вирионов при помощи специальной методики с применением детергентов. Может быть обнаружен в ядрах гепатоцитов при иммуноморфологическом исследовании биоптатов печени. В практической работе не определяется.
HBeAg — индикатор вирусной репликации, контагиозности крови и активности хронической HBV-инфекции. В отличие от HBcAg, прочно связанного с вирионным нуклеокапсидом, секретируется инфицированной клеткой. Имеется высокая корреляция между HBeAg-антигенемией и концентрацией вируса в крови.
Специфическая диагностика (выявление антител). Информативность анализа повышается при одновременном определении антител к HBV-антигенам. Изучение их спектра и динамики способствует уточнению диагноза, детализирует взаимоотношения между вирусом и хозяином, служит индикатором перенесенной HBV-инфекции и резистентности к ней.
Анти-HBs появляются после клинического завершения острого гепатита. Отражают окончание инфекционного процесса, свидетельствуют о перенесенной в прошлом инфекции (в том числе субклинической), служат маркером устойчивости к повторной HBV-инфекции. Отсутствуют у хронических носителей вируса.
Анти-HBc обнаруживаются в начальном периоде острого гепатита, а иногда в конце инкубации. Ранние анти-HBc относятся к классу IgM и выявляются на протяжении 3 мес. Их тестирование имеет принципиальное значение в HBsAg—анти-HBs негативном периоде острого гепатита (фаза окна). IgG анти-HBc появляются позже и, подобно анти-HBs, сохраняются в течение нескольких лет.
Анти-HBe появляются одновременно с анти-HBs, т.е. при завершении острого процесса. Это дополнительный критерий окончания инфекционного процесса и перенесенной в прошлом HBV-инфекции. При хроническом гепатите не выявляются.
Анти-HBx. Их появление совпадает с элиминацией вируса из организма. В практической диагностике не используются.
Анти-HBpol отражают активность хронического процесса, но не коррелируют с очищением печени от вируса при остром гепатите. В практической диагностике не используется.


Динамика специфических HBV-маркеров при остром гепатите В показана в таблице 2.
Cпецифическая профилактика. Для иммунопрофилактики гепатита В используется вакцина на основе рекомбинантного HBsAg. Разработаны вакцины, содержащие pre-S-антигены вируса гепатита В. От них ожидается более интенсивный и надежный ответ в группах риска, к которым, в частности, относятся медицинские работники. В экстренных случаях рекомендован анти-HBs-иммуноглобулин. Механизм действия таких препаратов связан с антителами, блокирующими HBsAg-зависимое прикрепление вируса к гепатоцитам.
В будущем ожидается создание вакцин для борьбы с персистентной инфекцией. Есть предложение конструировать их на основе производных кор-антигена, содержащих Т-эпитопы. Идея базируется на стимуляции Т-лимфоцитов, нацеленных против зараженных гепатоцитов, экспрессирующих на своей поверхности HBcAg.
Вирус гепатита D
Вирус гепатита D (HDV, дельта-агент) является мелким минус-РНК-вирусом, который способен поражать гепатоциты только в присутствии вируса гепатита В. Это говорит о его дефектности, которая проявляется в зависимости от вируса-помощника (HBV). По ряду признаков (устройство и репликация геномной РНК, сателлитизм, т.е. зависимость от вируса-помощника) HDV эволюционно близок вироидам растений и больше похож на «субвирус», чем на настоящий вирус.
HDV-геном (мельчайший среди известных РНК-вирусов человека — 1700 нуклеотидов) кодирует два или три полипептида, которые образуют чехол вокруг геномной молекулы и объединяются в понятие дельта-антиген (HDAg). Известно три генотипа HDV. Они имеют неодинаковое распространение (в России доминирует HDV-I), но лишены принципиальных антигенных различий: вирус представлен единственным HDAg-серотипом. Дельта-антиген не экспрессируется на поверхности инфицированных гепатоцитов и не принимает участия в реакциях Т-клеточного иммунитета. Антитела против HDAg (анти-HDAg) не обладают протективным эффектом.
Неожиданностью явилось открытие механизма транскрипции/репликации РНК у HDV: ее обеспечивает клетка-хозяин. До сих пор считалось, что клетки лишены РНК-зависимой РНК-полимеразной активности. Но вирус гепатита D каким-то образом перестраивает одну из клеточных ДНК-зависимых РНК-полимераз (полагают, что это РНК-полимераза II), заставляя ее выполнять необычную для себя роль РНК-зависимой РНК-полимеразы. Не исключено, что этот эффект может дополняться действием cубстратно измененной ДНК-полимеразы вируса-помощника (HBV). Дельта-вирус обладает еще одной почти уникальной особенностью: его РНК выполняет каталитическую (точнее аутокаталитическую) функцию. Она расщепляет мультимерный предшественник генома на мономерные фрагменты, которые используются для воссоединения в репликативный матрикс.
Хелперные функции вируса гепатита В до конца не выяснены. Одной из важнейших является предоставление дельта-агенту суперкапсида вместе с его главным компонентом — HBsAg. Результатом является образование вирулентного гепатотропного агента — вируса гепатита D. В этом, несомненно, задействована гепатотропность HBsAg, которая обеспечивает избирательное прикрепление вируса к гепатоцитам. Но есть и более глубокие механизмы взаимодействия дельта-агента и HBV. Они проявляются на стадии внутриклеточной репликации и важны для обоих вирусов. Полагают, например, что дельта-агент пользуется HBV-полимеразой с измененной субстратной специфичностью (см. выше), а его влияние на репликативные процессы HBV проявляется в том, что HBcAg вируса гепатита В и дельта-антиген никогда не сосуществуют в одной и той же клетке, т.е. их функции взаимоисключаемы. При одновременном инфицировании (HDV—HBV-коинфекция) активная репликация HDV часто приводит к подавлению репродукции HBV. Дельта-агент контролирует и собственную репликацию; эту роль выполняют субкомпоненты дельта-антигена c функциями позитивных и негативных регуляторов. Более того, HDV может инфицировать гепатоциты и без помощи вируса гепатита В. По крайней мере, его маркеры (HDAg и HDV-РНК) удается обнаружить в печени при полном отсутствии маркеров HBV. Этот вариант дельта-инфекции протекает бессимптомно, и лишь HBV-суперинфекция переводит ее в клинически манифестную форму. В первом случае можно говорить о персистенции вируса, во втором — о его стимуляции. HBV выступает в роли активатора, нарушая гомеостатический баланс между HDV и гепатоцитами. Впрочем, чаще бывает наоборот: дельта-инфекция наслаивается на персистентную HBV-инфекцию, обостряя течение процесса (HDV-суперинфекция).
Гепатит D и его возбудитель описаны в конце 70-х гг., т.е. значительно раньше, чем вирус гепатита С. Иными словами, алфавитная последовательность в этом случае формально нарушена. Вместе с тем, буквенное обозначение хорошо отражает историю открытия и утверждения дельта-инфекции как дискретной клинико-вирусологической категории. В 1977 г. итальянский вирусолог M. Rizetto при иммунофлюоресцентном исследовании биоптатов печени больных хроническим гепатитом В обнаружил в ядрах гепатоцитов, содержащих HBcAg, дополнительный антиген. Поначалу его расценили как неизвестный ранее компонент вируса гепатита В и обозначили (в алфавитной последовательности (!), т.е. вслед за HBcAg) буквой «дельта» — дельта-антиген, или HBdAg. Вскоре, однако, оказалось, что он не имеет отношения к HBV, а принадлежит самостоятельному вирусу, который стали называть дельта-агентом, а затем вирусом гепатита D (HDV). После открытия очередного возбудителя гепатитов (вируса гепатита С) утвердившуюся терминологию было решено не менять.
Гепатит D имеет глобальное распространение, хотя как и гепатит В регистрируется с неодинаковой частотой в различных регионах Земного шара: 0,1—30% от общего числа носителей HBsAg, в среднем около 5%. Зоны гиперэндемичности гепатитов В и D в основном совпадают (Южная Америка, Экваториальная Африка и др.). Подобно гепатиту В, это антропонозная кровяная инфекция. Единственным механизмом передачи служит парентеральная инокуляция возбудителя, который, высвобождаясь из поврежденных гепатоцитов, в огромных количествах накапливается в крови (до 1010 инфицирующих доз в 1 мкл для шимпанзе). По неизвестным причинам меньшее значение имеют половой и перинатальный пути передачи, в остальном эпидемиология дельта-инфекции не имеет существенных отличий от гепатита В.
Гепатит D развивается при одновременном заражении вирусом гепатита В (коинфекция) либо при инфицировании его носителей (дельта-суперинфекция). При коинфицировании клиника не отличается от картины острого гепатита В, хотя есть тенденция к более тяжелому течению и обострениям. Суперинфекция нередко ведет к тяжелым формам острого гепатита (число фульминантных случаев возрастает до 12%) и значительно (до 80%) повышает риск хронизации и ее исходов в цирроз. Вероятность развития первичной гепатомы не меняется.
В отличие от HBV, вирус гепатита D обладает прямой цитопатичностью, возможно потому, что HDV-РНК вызывает блокаду металлоорганических промоторов, играющих важную роль в клеточном метаболизме. Этому есть морфологические и цитохимические подтверждения, об этом же говорит и относительно короткий инкубационный период гепатита D (в среднем 35 дней, т.е. примерно в два раза короче, чем для гепатита В). Кроме того, HDV не экспрессирует своих эпитопов в мембране гепатоцитов, а потому не создает мишеней для цитотоксических Т-лимфоцитов.
Дельта-инфекция оставляет прочный иммунитет, но протективные антигены вируса не установлены. Не исключено, что, как и при гепатите В, эту роль выполняет HBsAg, который обеспечивает адсорбцию HDV на гепатоцитах.
Cпецифическая диагностика строится на выявлении в сыворотке дельта-антигена, HDV-РНК, анти-HDV IgM- и IgG-антител. Их профиль и динамика позволяют дифференцировать коинфекцию от суперинфекции, причем толчком к анализу служат высокие титры HBsAg. Это один из поводов для того, чтобы заподозрить наслоение дельта-инфекции. Практически значимым критерием хронического гепатита D является стойкое повышение анти-HDV IgM-антител на фоне отсутствия серологических маркеров репликативной HBV-инфекции (HBeAg, HBV-ДНК, анти-HBc IgM). Лишь у меньшей части больных выявляются признаки активной репликации обоих вирусов (HDV и HBV).
Вирус гепатита С
Вирусом гепатита С (HCV) инфицировано около 3% населения Земного шара, и фактически речь идет о пандемии, которая по масштабу в 5 раз превосходит зараженность вирусом СПИДа. После резкого уменьшения случаев гемотрансфузионного гепатита С (благодаря антивирусному контролю донорской крови) инфицированность поддерживается за счет парентеральных инъекций наркотиков и других процедур, связанных с повреждением кожи и слизистых оболочек. В большинстве заражаются молодые люди в возрасте 15—29 лет.
Инфицирование в 50—80% случаев ведет к персистенции вируса, которая служит причиной хронического гепатита и его фатальных осложнений — цирроза и первичного рака печени. В относительном измерении их вероятность не очень велика 1, но вследствие масштабности заражения число таких больных значительно. Современные методы диагностики позволяют с высокой точностью выявлять HCV-инфекцию и следить за ее эволюцией. Это служит основой для превентивных мероприятий и антивирусной терапии.
Вирусология. Открытие вируса гепатита С явилось беспрецедентным событием в вирусологии: он был идентифицирован не как вирусная частица (вирион), а как геномная молекула, фрагмент которой удалось транслировать после клонирования ДНК-копии. Лишь недавно появились инвитровые системы, поддерживающие репликацию генома и даже сборку HCV-подобных частиц, однако до сих пор единственной моделью, наиболее полно воспроизводящей HCV-инфекцию, является заражение шимпанзе.
HCV классифицирован как единственный вид рода Hepacivirus семейства Flaviviridae, куда входят такие (наиболее родственные ему) вирусы, как вирус желтой лихорадки, вирус денге и вирус гепатита G. Это мелкий (30—38 нм в диаметре) РНК-содержащий оболочечный вирус. Геном представлен линейной одноцепочечной плюс-РНК (примерно 9,6 кбайт). Она кодирует единственный полипротеин (3011 аминокислот), который расщепляется клеточными и вирусными протеазами на 3 структурных и 7 неструктурных белков. Структурные белки входят в состав нуклеокапсида (С-белок, от англ. сore — cердцевина) и оболочки/суперкапсида (Е1 и Е2, от англ. еnvelope — оболочка). Неструктурные белки не включаются в вирион. Они участвуют в репликации вируса, выполняя функции различных ферментов (протеазы, РНК-зависимой РНК-полимеразы, геликазы) (табл. 3). Для неструктурных белков приняты обозначения NS1 (p7), NS2, NS3, NS4A, NS4B, NS5A, NS5B (NS — от англ. nonstructural).
Таблица 3
Компоненты вируса гепатита С*
| Ген | Белок | Функция |
| С | р21/22 | Кор (нуклеокапсид) |
| Е1 | gp31 | Суперкапсид |
| Е2 | gp70 | Суперкапсид |
| NS1 | р7 | ? |
| NS2 | р23 | Протеаза |
| NS3 | p70 | Протеаза, геликаза |
| NS4 | р8 | Кофактор NS3 протеазы (NS4A) |
| NS5 | p68 | Кофактор репликативного комплекса (NS5A) |
| РНК-зависимая РНК-полимераза (NS5B) |
*Перечисление генов соответствует их 51®31 локализации; р — белки (от англ. protein), gp — гликопротеины (от англ. glycoproteins); цифры означают молекулярную массу (кДа). Функции р7 не известны; р7 может присоединяться к С-терминальной части Е2, определяя его изоформность.
На каждом из концов РНК имеется нетранслируемый участок — UTR (от англ. untranslated region; 329—341 нуклеотидов). UTR играют важную роль в репликации вируса, обеспечивая восприятие регуляторных клеточных сигналов. Наряду с неструктурными белками они являются потенциальными мишенями для антивирусной терапии.
Вариабельность. Для вируса гепатита С характерна высокая частота мутаций, связанных с ошибками в репликации РНК. По данным гомологии нуклеотидных последовательностей РНК предложено дифференцировать 6 основных генотипов и более сотни субтипов, обозначаемых латинскими буквами в порядке их открытия (1а, 1b, 2a, 2b, 2c, 3а и т.д.). Они неодинаково распространены в различных регионах Земного шара: некоторые — универсальны, другие циркулируют лишь в ограниченных географических зонах. В США и Западной Европе чаще встречаются субтипы 1а и 1b, за ним следуют субтипы 2 и 3. В России преобладает субтип 1b (50—80%). Для практических целей достаточно идентифицировать 5 наиболее распространенных субтипов — 1a, 1b, 2a, 2b и 3a.
Генетическая изменчивость HCV проявляется и по ходу инфекции. В зараженном организме вирус представлен популяцией близкородственных клонов с небольшими (1—2%) отличиями в структуре генома. Это так называемые квазивиды (англ. quasispecies). Они играют большую роль во взаимоотношениях с хозяином, обеспечивая уклонение от эффекторов иммунитета, расширение тканевого тропизма и повышение устойчивости к антивирусной терапии. Наиболее мутабельны области генома, кодирующие оболочечные белки, прежде всего Е2. Здесь находится один из двух гипервариабельных участков — HVR-1 (от англ. hypervariable region), который несет важную функциональную нагрузку, инициируя взаимодействие с клеточными рецепторами. Это делает его носителем протективных эпитопов, индуцирующих образование вируснейтрализующих антител. К сожалению, эффективность последних быстро падает, так как квазиспецифичность помогает вирусу ускользать от иммунного ответа — ускользающие мутанты (англ. escape mutants).
Эпидемиология. Источником заражения служат больные острыми и хроническими формами HCV-инфекции. Основное значение имеют лица с бессимптомным и малосимптомным течением. Будучи глобальной инфекцией, гепатит С неодинаково распространен в различных регионах Земного шара. Это доказывают результаты обследования доноров крови, среди которых процент серопозитивности (наличие HCV-антител) колеблется от 0,01—0,05% (Северная Европа, северные регионы США, Канада) до 6,0—28,0% (Египет). В России показатели анти-HCV колеблются от 0,6% (северные регионы) до 3,0% (Южная Сибирь, Дальний Восток) и в среднем составляют 1,5%. Если учесть, что у 3 из 4 серопозитивных лиц имеется виремия, то носителями активной НCV-инфекции являются около 2 млн. жителей России. Для сравнения те же показатели для США составляют 1,8% и 2,7 млн.
Формально эпидемиология гепатита С достаточно проста: вирус передается в основном при контакте с зараженной кровью и ее продуктами. По ретроспективным оценкам анти-HCV обнаруживаются в большинстве образцов донорской крови, переливание которой осложнилось гепатитом. У гемофиликов, получавших в прошлом многократные инъекции антигемофильных препаратов, антитела к HCV выявляются в 50—80% случаев. Высокий процент инфицированности (в 20 раз и более выше, чем у здоровых) имеют больные на постоянном гемодиализе. Вероятность нозокомиального заражения работников здравоохранения от случайных уколов шприцевыми иглами после инъекций HCV-инфицированным больным по разным данным колеблется от 0 до 10%.
Основную группу риска составляют лица, употребляющие парентеральные наркотики. Они играют ведущую роль в современной эпидемиологии гепатита С, определяя неодинаковую интенсивность эпидемического процесса в разных возрастно-половых и социальных категориях. Это происходит благодаря многократному использованию контаминированных вируссодержащей кровью игл и шприцев для внутривенного введения психотропных препаратов.
В 20—40% случаев причины заражения HCV остаются нераскрытыми, по крайней мере их не удается связать с известными факторами риска. Это говорит о возможности распространения вируса иными путями. Один из активно обсуждающихся механизмов — половые контакты. Такая возможность реальна, но маловероятна. Среди постоянных половых партнеров, один из которых хронически инфицирован HCV, риск заражения не превышает 1% в год. Некоторые авторы вообще отрицают присутствие вируса в семенной жидкости и вагинальном секрете, хотя это и не исключает половой передачи. Заражение может быть результатом мобилизации циркулирующего вируса через микротравмы слизистых оболочек при половом акте.
HCV содержится в слюне, но его концентрация скорее всего недостаточна для заражения.
Передача HCV от матерей — тоже редкое явление: вероятность контаминации плода и новорожденного инфицированными женщинами составляет 1—5%. Вирус, полученный в перинатальном периоде, длительно персистирует без сероконверсии (толерантность!); антитела могут появиться лишь в зрелом возрасте.
Пробелы в представлениях о естественном компоненте эпидемического процесса вынуждают говорить о криптогенной HCV-инфекции с неустановленным эпидемиологическим анамнезом (отсутствие факторов риска).
Патология. Большинство сведений о взаимоотношениях HCV с человеком получено из наблюдений за посттрансфузионным гепатитом, когда можно точно установить сроки заражения (рис. 5).

Критерием хронизации принято считать HCV-виремию продолжительностью более 6 мес. В большинстве случаев (70—80%) HCV-персистенция протекает при слабовыраженном поражении печени (латентное течение); отдаленный исход не известен и скорее всего благополучен, по крайней мере на ближайшие 20—30 лет. Это чаще наблюдается у женщин и при заражении в молодом возрасте. В остальных случаях (10—30%) хронический гепатит обретает агрессивный характер и в течение 10—20 лет завершается циррозом. У 20—30% таких больных со временем развивается рак печени. Реализации HCV-агрессивности способствуют следующие факторы: мужской пол, заражение в старшем возрасте (особенно после 50 лет), злоупотребление алкоголем, коинфицирование вирусами иммунодефицита человека (ВИЧ-1) и гепатита В. Значение HCV-генотипа не доказано. В частности, принадлежность к генотипу 1, с которым связана повышенная резистентность к антивирусной терапии, не имеет прогностического значения. Спонтанная элиминация вируса маловероятна.
Кроме гепатита при хронической HCV-инфекции возможны другие, внепеченочные осложнения. Они часто ассоциированы с аутоиммунными реакциями, отражая способность вируса к репликации в лимфоидной ткани, прежде всего в В-лимфоцитах. Большинство внепеченочных симптомов связано с продукцией криоглобулинов, обладающих свойствами ревматоидных факторов. Они обнаруживаются у половины лиц, инфицированных HCV, причем криопреципитаты обычно содержат HCV-антигены и анти-HCV-антитела. В 10—15% случаев развивается клиническая картина смешанной криоглобулинемии. Симптомы (слабость, артралгии, пурпура) формируются на основе васкулита. Наиболее тяжелые случаи протекают с поражением почек (мембранопролиферативный гломерулонефрит) и нервной ткани (периферические нервы, головной мозг). Сходным способом (синтез аутоантител, продукция иммунных комплексов) HСV включается в патогенез других, в том числе лимфопролиферативных заболеваний. Это дает право говорить о том, что хроническую HBV-инфекцию следует рассматривать не только как заболевание печени, но как системный процесс, в котором ведущую роль играет тканевой тропизм возбудителя.
Патогенез. Патогенетическая стратегия вируса гепатита С сводится к персистенции. Только закрепившись и поддерживая свою репликацию в организме, он представляет реальную опасность.
Для рецепции и эндоцитоза требуется взаимодействие между оболочечными (Е1 и Е2) белками вируса и мембранными молекулами (рецепторами) клеток. Центральная роль принадлежит связке CD81—Е2. Набор дополнительных рецепторов и лигандных Е1/Е2-сайтов может быть неодинаковым для гепатоцитов и других клеток, способных поддерживать репликацию HCV. Не исключено и то, что HCV-квазивиды отличаются по тканевому тропизму, используя особенности клеточных рецепторов.
Длительное пребывание HCV в организме определяется его способностью выживать в условиях достаточно напряженного и разнообразного иммунного ответа. Антитела не защищают от HCV-персистенции и не обеспечивают надежного иммунитета против реинфекции. Скорее всего, это связано с антигенной вариабельностью (даже гипервариабельностью) оболочечных белков, благодаря которой минорные (т.е. по началу немногочисленные) иммунорезистентные вирусные клоны ускользают от нейтрализующих антител, обретая лидерство в патологическом процессе. Замечено, что вируснейтрализующие антитела, продуцируемые в раннем периоде хронического гепатита, не эффективны против HCV-изолятов, полученных от тех же больных в более поздние сроки. Имеет значение и то, что многие HCV-вирионы ассоциированы с сывороточными липопротеинами (b-липопротеины низкой и очень низкой плотности), которые экранируют вирусные антигены, защищая HCV от антител. Одновременно это создает условия для дополнительного взаимодействия вирионов с клетками — через рецепторы для липопротеинов низкой плотности.
Не всегда результативны и эффекторы Т-клеточного иммунитета. Несмотря на то, что структурные и неструктурные HCV-белки содержат множество Т-эпитопов, выступающих в качестве мишеней для хелперных (CD4+) и цитотоксических (CD8+) Т-лимфоцитов, Т-зависимая элиминация вируса часто бывает неполной. Кроме антигенной вариабельности, имеют значение сродство пептидов к презентирующим их молекулам HLA, сокращение внутриклеточного пула вируса до уровня, не воспринимаемого Т-лимфоцитами, вирусиндуцированное ослабление экспрессии HLA на инфицированных клетках, а также клональная анергия Т-лимфоцитов, связанная с действием так называемых антагонистических Т-эпитопных вирусов-мутантов. Все эти механизмы, которые широко обсуждаются в связи с феноменом вирусной персистенции, повышают устойчивость HCV к эффекторам иммунитета, содействуя стабилизации инфекции в организме.
Еще один механизм, способствующий выживанию HCV, связан с его антиинтерфероновой активностью. Это, в частности, объясняется блокадой клеточной протеинкиназы, которая обеспечивает один из антивирусных эффектов интерферона — опережающее подавление синтеза вирусных белков за счет торможения фактора элонгации-2. Участок NS5A, ответственный за этот эффект, неодинаково активен у различных генотипов вируса, определяя разную степень рефрактерности к антивирусной терапии. В наибольшей степени это выражено для штаммов генотипа.
Персистенции HCV содействуют внепеченочные резервуары инфекции, прежде всего в лимфоидной ткани. Вирус находит здесь не только пассивное убежище. Это дополнительная зона реализации его агрессивного потенциала. Подвергаясь репликации, вирус вызывает функциональные перекосы лимфоцитов, которые ведут к развитию заболеваний иммунной системы (см. выше) и ослабляют ее вирусэлиминирующий потенциал. Это, в частности, зависит от нарушения баланса регуляторных цитокинов. Преобладание Th2-цитокинов при остром НCV-гепатите ассоциируется с ослаблением эффекторного звена Т-клеточного иммунитета, содействуя вирусной персистенции и хронизации инфекции. Напротив, доминирование Th1-цитокинов коррелирует с тенденцией к выздоровлению и элиминацией возбудителя.
Механизмы HCV-зависимого поражения гепатоцитов остаются неясными. Прямая цитопатичность реальна, но, безусловно, не является главной причиной. Существенная роль принадлежит иммунологическим механизмам повреждения, прежде всего цитодеструктивным реакциям антивирусных Т-лимфоцитов, нацеленных против клеток, несущих вирусные пептиды в комплексе с молекулами HLA. Процент клеток, содержащих РНК-HCV, колеблется в широких пределах (от 4,8 до 87,6%), определяя вероятный масштаб Т-зависимой агрессивности. Патология возникает, когда гибель клеток не обеспечивает полноценной элиминации вируса из-за ускользания HCV-мутантов от иммунного пресса.
Повышенный риск развития гепатоцеллюлярной карциномы, который воспринимается как один из грозных символов хронического гепатита С, не имеет четких объяснений. Впрочем, это едва ли возможно, если учесть, что канцерогенез — многоступенчатый процесс, в котором задействованы разнообразные факторы и механизмы. В отличие от вируса гепатита В, HCV-геном не вступает в интеграцию с ДНК зараженных клеток. Это исключает один из принципиальных механизмов онкогенности — инсерционный мутагенез, побуждая искать другие объяснения. По одной из версий, онкогенность HCV связана с производными кор(С)-протеина, которые, взаимодействуя с регуляторными белками клетки-хозяина, влияют на экспрессию генов, ответственных за регуляцию роста и апоптоза клеток. Однако редкое развитие гепатокарциномы в нецирротической печени исключает универсальность данного механизма, позволяя думать об опосредованном характере гепатоканцерогенеза. Допускают, например, что рак развивается в результате спонтанного мутагенеза гепатоцитов, побуждаемых к перманентной регенерации в условиях хронической вирусиндуцированной гибели печеночной паренхимы. Этому содействует повышенная чувствительность клеток к вторичным (эндогенным и экзогенным) мутагенам, которые особенно опасны для одноцепочечной ДНК, временно образующейся по ходу клеточной репликации.
Иммунитет. Не обеспечивая полной элиминации вируса, иммунная система существенно влияет на развитие HCV-инфекции. Выше об этом говорилось в связи с иммунологически зависимыми механизмами патогенеза хронического гепатита С и внепеченочных осложнений. Но иммунный ответ может быть и достаточно эффективным. Об этом свидетельствуют вероятность полного очищения от вируса (по разным данным, у 20—50% остро инфицированных), а также многолетняя стабилизация инфекции на умеренном уровне у большинства зараженных.
Характерный признак гепатита С — отсроченная сероконверсия. Первые анти-HCV-антитела удается обнаружить не ранее 5 нед после инфицирования (переливание зараженной крови), но сроки их появления могут быть и гораздо длиннее (30—50 нед). Спектр антител достаточно сложен, отражает эпитопную поливалентность HCV-антигенов. Динамика антител к различным антигенам (точнее их эпитопам) не одинакова, что имеет диагностическое значение.
Вируснейтрализующие антитела ограничивают инфекцию, но пропускают мутантные клоны (квазивиды) с обновленными В-эпитопами. Это тем более вероятно, поскольку протективные В-эпитопы сосредоточены в гипервариабельных зонах оболочечных антигенов, прежде всего Е2. Между титрами вируснейтрализующих антител и исходами острого гепатита нет корреляции, а у лиц, спонтанно выздоровевших от острого гепатита, их содержание невелико. Образование нейтрализующих антител продолжается и при хронической инфекции, но их эффективность здесь еще ниже. В опытах на шимпанзе антитела предохраняют от реинфекции своим штаммом, но не защищают от заражения гетерологичными штаммами даже идентичного генотипа. У детей с врожденной гипогаммаглобулинемией течение хронической HCV-инфекции не имеет заметных особенностей.
Это определило интерес к реакциям Т-клеточного иммунитета, которым принадлежит главная роль в элиминации и длительном сдерживании HCV. Основой служит относительный консерватизм протективных Т-эпитопов, представленных на всех HCV-белках — cтруктурных и неструктурных. Принципиально, что вирусинфицированные клетки становятся мишенями для Т-лимфоцитов до созревания новых вирусных частиц. Высокая сенсибилизация Т-лимфоцитов (CD4+ и CD8+) к HCV-антигенам характерна для острого гепатита С, который завершается элиминацией возбудителя, но не для больных с исходом в хроническую инфекцию. В последнем случае реакции выражены слабее и явно недостаточны для эрадикации вируса. Задача решается здесь частично, иногда с потерями для хозяина: стабилизируя HCV-персистенцию, Т-лимфоциты поддерживают хроническую инфекцию, включаясь в патогенез (см. выше).
Высокая изменчивость HСV затрудняет создание профилактической и лечебной (для ликвидации персистентной инфекции) вакцин. Новые идеи связаны с Т-вакцинами, которые в отличие от традиционных вакцин (нацеленных на образование антител) рассчитаны на избирательную активацию антивирусных Т-лимфоцитов. С этой целью кроме HCV-пептидов планируется использовать генетические (ДНК) вакцины, которые основаны на рекомбинантных молекулах ДНК — бактериальные плазмиды со встроенными HCV-генами. Попав в клетки, они подвергаются экспрессии с образованием антигенных пептидов, которые включаются в индукцию иммунных реакций.
Специфическая диагностика. Диагноз HCV-инфекции базируется на выявлении специфических серологических маркеров. Это важно не только для дифференциальной диагностики гепатита С, но и для суждения о стадии заболевания, для прогнозирования его течения, оценки активности процесса и эффективности противовирусной терапии.
Принципиальное значение имеет генодиагностика, основанная на детекции РНК-HCV. Недавно появилась возможно для обнаружения циркулирующих HCV-антигенов.
Анти-HCV-антитела. Выявление анти-HCV-антител проводят при помощи твердофазного иммуноферментного анализа (ИФА) на основе комплекса структурных и неструктурных вирусных пептидов. В настоящее время применяются диагностические системы 3-го поколения, которые построены из рекомбинантных и/или синтетических фрагментов структурных и неструктурных HCV-белков (С, NS3, NS4 и NS5). Внедрение таких диагностикумов позволило сократить сроки первичного выявления анти-HCV при острой инфекции, существенно повысить чувствительность и специфичность реакции. Первые антитела (обычно анти-С, анти-NS3, анти-NS5) удается обнаружить через 20—150 дней (в среднем через 50 дней). Частота выявления анти-HCV среди РНК-HCV-позитивных образцов крови приближается к 100%. На фоне хронической инфекции антитела выявляются постоянно, а после элиминации вируса сохраняются (прежде всего анти-С) в течение 4—8 лет и более. Поэтому формальное наличие анти-HCV не всегда говорит о присутствии вируса в организме и не позволяет судить об активности процесса.
Для суждения о вирусной нагрузке, активности HCV-репликации, риске хронизации, разграничении острого и хронического гепатита, длительности инфекционного процесса и степени поражения печени предложено использовать определение спектра антител к различным HCV-пептидам, а также наблюдать за их качественной и количественной динамикой. Это дает ориентировочную информацию, которая требует подтверждения другими методами.
Определенный резонанс получило предложение о дифференцированном определении IgM-антител против С(кор)-антигена. Вопреки ожиданиям IgM-ответ в острой фазе гепатита С не следует классическому пути антителообразования: IgM анти-HCV могут выявляться одновременно и даже позднее, чем анти-HCV класса IgG. Поэтому обнаружение IgM анти-HCV не может быть использовано как маркер острой HCV-инфекции. Вместе с тем длительность циркуляции анти-кор-IgM (3—5 мес) является фактором, прогнозирующим персистентную инфекцию, а их появление при хроническом гепатите С свидетельствует о реактивации вируса, т.е. об обострении процесса. Этому соответствует корреляция между темпами снижения анти-кор-IgM и эффективностью противовирусной терапии.
Несмотря на высокую специфичность, современные ИФА-системы не гарантированы от гипердиагностики, т.е. от ложноположительных результатов. Для их исключения требуется динамическая оценка на основе индикации анти-HCV к дискретным антигенам (подтверждающие, или конфирмативные тесты). Для этого используют ИФА-анализ в реакциях с раздельно сорбированными на планшетах HCV-пептидами или иммуноблоттинг.
Наряду с ложноположительными возможны и ложноотрицательные результаты, когда анти-HCV-антитела не удается обнаружить, несмотря на присутствие вируса в организме. Известны по меньшей мере три подобных ситуации:
- начальный период заболевания (серонегативная фаза острого гепатита — фаза окна);
- больные, получающие иммунодепрессанты (они могут быть длительно инфицированы без сероконверсии);
- заражение некоторыми HCV-генотипами (прежде всего 3 и 4).
В таких случаях требуются прямые доказательства инфицированности.
РНК-HCV. Выявление РНК-НCV считается «золотым стандартом» в диагностике гепатита С. Наиболее широко используется классический вариант полимеразной цепной реакции (ПЦР) после обратной транскрипции HCV-РНК в комплементарную ДНК. Таким путем удается следить за вирусемией, а также судить о присутствии HCV в печени и других тканях. Определение циркулирующей HCV-РНК наиболее часто применяют для подтверждения антительных тестов, раннего диагноза острого гепатита (РНК-HCV удается обнаружить на 7—21-й день после инфицирования, т.е. задолго до появления первых антител), мониторинга перинатального заражения и контроля за эффективностью антивирусной терапии.
В целом данные HCV-РНК-вирусемии хорошо коррелируют с обнаружением анти-HCV-антител. Положительные результаты ПЦР в сочетании с негативными реакциями на анти-HCV характерны для серонегативного периода острого гепатита (фаза окна). Негативные показатели на фоне положительных антительных тестов могут быть следствием ложной позитивности последних либо низкой (не улавливаемой в ПЦР) концентрации вируса в крови. Повторные анализы могут дать положительный результат, отражая активацию вируса в гепатоцитах или во внепеченочных резервуарах.
Кроме слежения за вирусемией, HCV-ПЦР применяется для обнаружения вируса в биоптатах печени. Это дает более полную информацию об эволюции инфекционного процесса, так как вирус способен персистировать в гепатоцитах, не выходя в кровь или присутствуя в ней в субпороговых концентрациях. Это чаще происходит на ранних этапах инфекции, но наблюдается и при хроническом HCV-гепатите.
HCV-антигены. Принципиальная возможность выявления структурных и неструктурных HCV-белков была установлена вскоре после открытия вируса при иммунофлюоресцентном изучении биоптатов печени больных хроническим гепатитом С и шимпанзе, зараженных HCV. Определение HCV-антигенов в сыворотке из-за их низкого содержания долго не удавалось. Лишь недавно разработаны методические подходы для иммуноферментного выявления С(кор)-белка в крови и организовано производство первых коммерческих тест-систем. Их внедрение в практику позволит решать спорные вопросы диагностики на более экономичной основе, чем определение HCV-РНК.
Принципы антивирусной терапии. Стратегия терапии хронического гепатита С направлена на ликвидацию хронической (персистентной) HCV-инфекции. Современная тактика предусматривает длительное комбинированное лечение интерфероном-альфа и рибавирином (синтетический аналог нуклеозидов). Испытание ряда рекомбинантных интерферонов не выявило их преимуществ. Предложен интерферон, химически связанный с полиэтиленгликолем (ПегИнтрон), что увеличивает период полураспада и продолжительность действия препарата.
По результатам устойчивого вирусологического ответа в рандомизированных группах больных (отсутствие РНК-HCV через 6 мес и более после отмены препаратов) эффективность комбинированной терапии составляет около 50%. Некоторые авторы отстаивают целесообразность антивирусного лечения в латентной фазе хронической HCV-инфекции, но другие не видят в этом смысла. Компромиссом служит признание целесообразности лечения HCV-РНК-позитивных лиц без поражения печени, но с признаками снижения качества жизни.
Склонность к высокой изменчивости HCV проявляется и в селекции клонов, устойчивых к антивирусной терапии. В сочетании с природной резистентностью HCV (прежде всего генотипа 1) к интерферону это создает дополнительные сложности в эрадикации персистентной инфекции, предрасполагая к вирусологическим рецидивам. В перспективе можно рассчитывать на препараты, которые будут действовать на дополнительные вирусные мишени, в частности блокировать вирусспецифические ферменты.
Вирус гепатита Е
Вирус гепатита Е (HEV) обнаружен М.С. Балаяном и соавт. (1983) в фекалиях больных острым гепатитом, у добровольца, зараженного экстрактом из этих фекалий, и в испражнениях экспериментально инфицированных обезьян (cynomolgus macaques). Культивировать in vitro вирус не удается, но его пассируют на приматах.
Под электронным микроскопом вирион HEV выглядит как безоболочечная частица, с кубическим типом симметрии, 32—34 нм в диаметре, с шипами и вдавлениями на поверхности. Геном представлен одноцепочечной плюс-РНК, размером 7,6 кбайт, с тремя открытыми рамками считывания (ОРС). Гены самой большой из них, ОРС1, кодируют неструктурные белки, связанные с репликацией вируса. В ней имеется гипервариабельный участок, который по последовательности нуклеотидов отличается у разных штаммов, отражая потенциальную изменчивость вируса. Вторая по размеру, ОРС2 кодирует структурные белки капсида, содержащие протективные эпитопы. Функции белков, кодируемых ОРС3, не известны, но они, подобно продуктам ОРС2, включают эпитопы, реагирующие с антителами сывороток больных гепатитом Е и реконвалесцентов.
По ультраструктуре и физико-химическим свойствам вириона вирус гепатита Е напоминает калицивирусы — семейство, близкое пикорнавирусам (он, в частности, похож на вирус Norwalk, один из возбудителей вирусной диареи человека). Однако детальное изучение генома показало невозможность включения HEV в семейство Caliciviridae. Сегодня он относится к «неклассифицируемым» вирусам.
Гепатит Е является энтеральной инфекцией, т.е. передается при помощи фекально-орального механизма. Подобно гепатиту А, пик выделения вируса с фекалиями падает на доклинический (инкубационный) период, что затрудняет проведение профилактических мероприятий. Сообщения о повышенных титрах антител против HEV среди групп риска по контакту с чужой кровью противоречивы, хотя известны случаи, когда вирусемия продолжается довольно долго (45—112 дней), создавая теоретическую возможность парентеральной передачи возбудителя.
Распространение гепатита Е ограничено, главным образом, развивающимися и экономически отсталыми странами тропического и жаркого поясов. Первая вспышка гепатита Е (30000 случаев острой желтухи) была зарегистрирована в Нью-Дели (Индия) в 1955—1956 гг. Она сопутствовала наводнению реки Ямуна, в которую стекались сточные воды. На основании эпидемиологических (фекально-оральный механизм передачи) и клинических (отсутствие хронизации) данных был заподозрен гепатит А, но ретроспективный анализ законсервированных сывороток поставил вопрос о новом возбудителе. Позже вспышки водного гепатита, лишенного маркеров HAV-инфекции, наблюдались в Пакистане, Юго-Восточной Азии, Африке, Мексике и Средне-Азиатских республиках бывшего СССР. Во время эпидемии, охватившей в 1986—1988 гг. китайскую провинцию Хиньянг-Юхар, заболело более 100000 человек.
Всего эпидемии гепатита Е описаны в 29 странах мира. Они обычно сопутствовали дождливым сезонам, наводнениям и были связаны с употреблением воды, загрязненной фекалиями (сточными водами). Контактно-бытовая передача также возможна и рассматривается как самостоятельный фактор риска. Но в отличие от гепатита А этот путь имеет меньшее значение, что, по-видимому, связано с относительно низкой болезнетворностью вируса. Это требует высокой дозировки инфекта, вероятность которой особенно реальна при использовании контаминированной воды. Обычно поражаются лица молодого и среднего возраста (15—40 лет). Дети до 15 лет болеют значительно реже, инфекция у них чаще протекает латентно. Это означает, что бессимптомная инфекция либо не обеспечивает иммунитета, либо он непродолжителен. Первое впечатление о том, что мужчины болеют чаще женщин, не подтвердилось.
Гепатит Е не только вызывает эпидемические вспышки, но и является серьезной проблемой спорадической заболеваемости. В регионах, эндемичных по гепатиту Е, он составляет более половины всех случаев острого вирусного гепатита (в Индии, например, ежегодно регистрируется около 2 млн. больных гепатитом Е). По клинико-эпидемиологическим признакам спорадический гепатит Е не отличается от эпидемического варианта, хотя фекально-оральный механизм заражения в этом случае чаще реализуется контактно-бытовым путем. Интригующей и загадочной остается повышенная чувствительность к HEV беременных женщин. Если общее число летальных исходов спорадического гепатита в эндемичных регионах составляет 0,5—4%, то для беременных женщин (особенно в 3-м триместре) этот показатель достигает 20%.
До сих пор не зафиксировано ни одной вспышки гепатита Е в развитых странах. Единичные, серологически документированные случаи заболевания в США и Европе в большинстве были привозными, т.е. болели лица, возвратившиеся из поездок в регионы, эндемичные по гепатиту Е. По результатам обследования доноров крови, серопозитивность по HEV среди здорового населения Европы составляет 1—2%, хотя при более жесткой оценке, направленной на исключение ложноположительных результатов, этот показатель, по-видимому, ниже.
Инкубационный период гепатита Е составляет 2—9 (в среднем 6) нед. Клиника напоминает картину гепатита А и не отличается от других острых вирусных гепатитов. Общие симптомы включают лихорадку, абдоминальные боли, артралгии, тошноту, рвоту, гепато- и спленомегалию; в сыворотке повышается уровень трансаминаз. Символом заболевания является желтуха, но и безжелтушные формы — не редкость. Возможна субклиническая инфекция, но процент бессимптомных форм не известен, так как серологическое тестирование проводилось в ограниченных масштабах.
Обычно гепатит Е протекает доброкачественно и заканчивается самоизлечением. Пер-систенция вируса и хронизация процесса не встречаются. Редким осложнением является фульминантный гепатит. По неизвестным причинам его вероятность возрастает у беременных женщин, особенно в 2—3-м триместре.
В преджелтушной стадии вирус обнаруживается в сыворотке и фекалиях. Согласно экспериментальным данным, после репликации в печени он, как и вирус гепатита А, вторично попадает в пищеварительный тракт из желчевыводящих путей. С развитием гепатоцеллюлярного некроза (это совпадает с повышением уровня сывороточных трансаминаз и пиком клинической симптоматики) размножение вируса подавляется, и он исчезает из желчи и фекалий. Появление антител отражает завершение патологического процесса в печени и начало выздоровления. Отсутствие сероконверсии служит плохим прогностическим симптомом, говоря о возможности рецидива.
Появление IgM-антител опережает пик АлАТ-активности; они снижаются при выздоровлении и исчезают в среднем через 5 мес после клинико-биохимического разрешения процесса. Образование IgG-антител начинается вскоре после обнаружения IgM-антител и нарастает по мере реконвалесценции. Первое впечатление о том, что IgG-ответ при гепатите Е недолговечен, оказалось неверным: высокие титры IgG-антител сохраняются на протяжении 1—4,5 лет после выздоровления.
Диагноз базируется на выявлении IgM-антител против пептидов, кодируемых ОРС2 и ОРС3; обнаружение суммарных антител без разграничения их класса менее информативно. Разработан и конфирмативный тест, основанный на иммуноблоттинге с рекомбинантными HEV-антигенами. Широкое внедрение серологических тестов только начинается. До сих пор не известны масштаб антигенной вариабельности вируса и то, в какой степени она может повлиять на полноценность иммунодиагностики. Не исключено, например, что серологически не расшифрованные случаи фульминантного гепатита связаны с антигенными мутантами вируса, ускользающими от иммунотипирования в стандартных тест-системах. О такой возможности говорят наличие в геноме HEV (ОРС1) гипервариабельного участка, а также наблюдения о фенотипических особенностях HEV-штаммов.
Как и при гепатите А, в раннюю фазу болезни в крови примерно 75% больных можно выявить HEV-РНК при помощи полимеразной цепной реакции.
При профилактике следует учитывать, что главным фактором риска для жителей развитых стран является посещение территорий, эндемичных по гепатиту Е.
Превентивным эффектом для туристов, посещающих эндемичные регионы, обладает местный иммуноглобулин; препараты, полученные в развитых (не эндемичных по гепатиту Е) странах не эффективны, так как не содержат анти-HEV-антител. Ожидается создание вакцины на основе рекомбинантного полипротеина ОРС2. Пока лучшей профилактикой является соблюдение правил личной и общественной гигиены, базирующихся на представлениях об экологии возбудителя и эпидемиологии HEV-инфекции.
Вирусы, претендовавшие/претендующие на избирательную гепатотропность
Несмотря на значительные успехи в изучении инфекционных гепатитов, определенное число их остается этиологически не расшифрованным. Это служит поводом для поиска гепатотропных вирусов, которые после открытия вируса гепатита С получили новое генетическое содержание. Обычно используют генно-инженерную биотехнологию, обеспечивающую избирательное размножение (амплификацию) in vitro нуклеотидных последовательностей ДНК и РНК с последующим клонированием в бактериальных или эукариотических клетках. Это позволяет синтезировать рекомбинантные антигены и геномные зонды, которые можно использовать для индикации вирусной инфекции, не имея вируса. Иными словами, открытие начинается не с вириона, а с его геномной молекулы, на основе которой искусственно воссоздаются элементы фенотипа.
Вирус(ы) гепатита F
Понятие «гепатит F» остается терминологическим курьезом. Им неоднократно пользовались для обозначения этиологически не расшифрованных случаев гепатита парентерального и энтерального генеза. Ни одна из этих работ не получила развития и/или подтверждения, но вирусологи американской фирмы Genelabs, открывшие в 1995 г. новый вирус парентерального гепатита, посчитали позицию «F» занятой и назвали обнаруженный ими агент вирусом гепатита G.
Вирус гепатита G
Предположение о двух разных вирусах, вызывающих парентеральный гепатит ни А—В, высказано более 20 лет назад. Гипотеза переросла в убеждение после открытия вируса гепатита С, когда оказалось, что у некоторых больных острым и хроническим гепатитом, подозревавшихся в парентеральных контактах, не удается обнаружить маркеры HBV и HCV. HBV- и HBC-негативные случаи регистрируются и среди больных «бытовым» гепатитом.
Геном вируса гепатита G был независимо клонирован в двух лабораториях, работающих под эгидой крупнейших коммерческих фирм — Abbot и Genelabs. Специалисты лаборатории Abbot занимались изучением вируса, который в конце 1960-х гг. был трансмиссирован обезьянам (тамаринам) от 34-летнего хирурга на третий день желтухи. Это произошло в Африке, а так как больной имел инициалы G и B, гепатит получил название GB-гепатит. Он не имел маркеров гепатитов А и В, а после открытия возбудителей гепатитов С и Е стало ясно, что его причиной был неизвестный вирус (или вирусы). В сыворотке зараженных тамаринов, на которых пассировался этот агент, была обнаружена уникальная РНК, получившая условное название GB-вирус (GBV). Oказалось, что GB-геном не однороден и представлен двумя дискретными (хотя и родственными) РНК-последовательностями — GBV-A и GBV-B. На их основе были получены рекомбинантные белки, дававшие положительные антительные реакции с сыворотками некоторых больных гепатитом ни А—Е и практически здоровых людей.
Однако претензии авторов на открытие новых гепатотропных вирусов человека встретили возражения. Их главным доводом было то, что гипотетические вирусы являются не вирусами человека, а обезьяньими вирусами, которыми в результате множества пассажей был разбавлен исходный GB-агент. В таком случае присутствие анти-GBV-A- и/или анти-GBV-B-антител у человека следовало объяснить антигенным родством этих вирусов с неизвестным (ни А—Е) вирусом человека. В связи с этим была предпринята попытка прямого обнаружения GBV-РНК в сыворотке анти-GBV-A- и анти-GBV-B-позитивных людей с факторами риска по парентеральному гепатиту. В ходе этих исследований были обнаружены уникальные РНК-последовательности, которые могли принадлежать лишь новому, не известному ранее вирусу. Генетически он был ближе к GBV-A, чем GBV-B и, подобно этим вирусам, имел некоторое сходство с вирусом гепатита С. Авторы временно назвали новый вирус GBV-C и отнесли его к флавивирусам (т.е. к тому же семейству, куда включен вирус гепатита С и два других GB-вируса), высказав предположение, что он является одним из возбудителей гепатита ни А—Е.
Авторский коллектив, ядро которого составили специалисты из Genelabs, прошел более короткий путь. При помощи оригинальной модификации полимеразной цепной реакции им удалось выделить РНК гипотетического вируса из крови обезьян (тамаринов), зараженных плазмой больных гепатитом ни А—Е. Клонированную РНК определили как вирус гепатита G (HGV), так как к этому моменту уже высказывались предположения о вирусе гепатита F (см. выше). На основании секвенирования (определения последовательности) нуклеотидов HGV был отнесен к семейству Flaviviridae. Было установлено его отдаленное генетическое родство с вирусом гепатита С и высокое — с GBV-C. Считается, что HGV и GBV-C — один и тот же вирус, и его часто обозначают как HGV/GBV-C.
Геном вируса гепатита G представлен плюс-РНК, которая кодирует полипротеин, состоящий примерно из 2900 аминокислот. По структурно-функциональной организации HGV-геном похож на РНК флавивирусов. К 5′-концу прилегают структурные гены, кодирующие белки сердцевины (соr) и оболочки (env). Неструктурные гены (для геликазы, протеиназы, РНК-зависимой РНК-полимеразы) расположены на 3′-конце РНК; они имеют сходство с GBV-А, GBV-B и HCV. Структурные гены не имеют ничего общего с GBV-B и HCV и лишь отдаленно напоминают GBV-A. Различные изоляты HGV имеют высокий процент гомологии (90—97%) с эталонным HGV (PNF2161), хотя, подобно вирусу гепатита С, масштаб этой гомологии неодинаково распределен по длине РНК-генома: 5′-нетранслируемый участок отличается высокой консервативностью, тогда как гены, кодирующие гипотетические структурные белки, значительно различаются у разных изолятов, и по этому признаку вирус может быть разделен на несколько генотипов.
Открытие HGV явилось результатом поиска вирусов, которые должны заполнить негативную нишу парентеральных гепатитов. Исходным материалом для выделения нового вируса послужила кровь гепатитных больных, и с этой точки зрения потенциальную патогенность HGV можно считать, казалось бы, доказанной. Однако о вероятности ее реализации сегодня можно говорить с большим сомнением. Присущее гепатиту С прогрессирование процесса с развитием хронического гепатита и его осложнений для HGV не характерно, несмотря на персистенцию вируса, диагностируемую по длительной РНК-вирусемии. HGV-РНК обнаруживается у 1—5% нормальных доноров крови. При повышенных показателях аминотрансфераз (АлАТ) носительство HGV возрастает, хотя, по другим данным, доноры с нормальным и повышенным уровнем АлАТ не отличаются по HGV-РНК-вирусемии.
Следует помнить, что гепатоциты могут быть латентно инфицированы практически безвредными вирусами, открытие которых грозит засорением гепатитного алфавита. В частности, нет полной уверенности в том, что HGV и родственные ему вирусы не будут балластом в классической гепатологии. Большинство авторов вообще отрицают его избирательную гепатотропность, полагая, что HGV не реплицируется в гепатоцитах. С этой точки зрения гепатит G такая же случайность, как поражение печени другими вирусами, например цитомегаловирусом. Персистентная HGV-моноинфекция, приближаясь к здоровому носительству, а острая клиника для HGV нетипична.
Вирус ТТ
В 1997 г. появилось сообщение японских авторов об открытии нового вируса, TTV, ассоциированного с поражением печени после гемотрансфузии. ТТV обнаружен как фрагмент геномной молекулы, в данном случае — специфической последовательности ДНК. Вирус назван по инициалам больного, в сыворотке которого он был впервые идентифицирован. За несколько лет, прошедших с момента открытия, TTV подвергся интенсивному изучению, в том числе как вероятный возбудитель гепатита человека. Сразу заметим, что первое впечатление о его патогенетической значимости не подтвердилось, хотя бы потому что ТТV-вирусемия широко представлена у здоровых людей. Впрочем, именно это вызывает дополнительный интерес к вирусу, так как подобное явление не имеет прецедентов.
TTV предположительно включен в семейство Circoviridae. Это означает, что он содержит несегментированный циркулярный геном, представленный однонитевой ДНК размером около 2,6 кбайт, вирион (30—50 нм в диаметре) лишен липидной оболочки, капсид имеет кубическую симметрию.
ДНК обладает небольшой генетической емкостью, объединяя три открытых рамки считывания (одна из них кодирует неструктурный белок, задействованный в вирусной репликации) и нетранслируемый участок (около 1,2 кбайт), воспринимающий регуляторные сигналы от клетки. В нетранслируемом фрагменте имеется множество инвертированных повторов, предполагающих вероятность внутригеномных перестроек и, следовательно, спонтанных мутаций. Капсид построен из единственного структурного белка — VP1. Если забыть про циркулярность ДНК, Circoviridae напоминают парвовирусы, к которым поначалу был отнесен ТТV. Впрочем, это сходство носит чисто формальный характер и относится лишь к размеру, строению капсида и высокой устойчивости во внешней среде.
Нынешняя таксономика ТТV удовлетворяет не всех вирусологов. Дело том, что ранее обнаруженные цирковирусы животных (возбудители болезни клюва и перьев попугаев, мультисистемной патологии поросят и анемии цыплят) имеют лишь слабую гомологию ДНК между собой и с ТТV. На этом основании, а также из-за различий в размерах вириона и генома, ТТV предложено считать первым представителем нового семейства вирусов — Circinoviridae. Несколько иначе выглядит позиция, базирующаяся на недавнем открытии еще одного вируса человека — TTV-подобного минивируса. Его геном занимает промежуточное положение между TTV и вирусом анемии цыплят, который по организации генома ближе всего стоит к TTV. Все они претендуют на объединение в самостоятельный таксон — семейство Paracircoviridae. Не исключено, что сюда попадет и вирус SEN, недавно обнаруженный в крови человека (см. ниже).
По аналогии с цирковирусами животных можно полагать, что репликация TTV происходит в ядре и зависит от белков клетки-хозяина, экпрессируемых в S-фазе клеточного цикла. Вирионы собираются в цитоплазме и высвобождаются путем цитолиза.
TTV-изоляты характеризуются высокой генетической неоднородностью. Это позволяет дифференцировать более десятка генотипов, а внутри них —различные субтипы. Практически вся гетерогенность связана с кодирующей частью генома, нетранслируемая область высоконсервативна (более 90% идентичности у разных штаммов). Удивительно, но цирковирусы животных гораздо менее вариабельны. Относительная гомогенность характерна и для большинства других ДНК-вирусов, в том числе для вирусов человека, имеющих однонитевую ДНК (парвовирус В19). Одной из причин может быть то, что циркуляция TTV и TTV-подобных вирусов среди нечеловекообразных приматов и домашних животных создает вероятность перекрестного обмена генами, который способствует селекции мутантных клонов. Не исключены и генетические рекомбинации при множественном заражении человека разными генотипами TTV и родственными вирусами животных. Нельзя, наконец, забывать об иммунном прессе, который должен испытывать вирус, длительно поддерживающий продуктивную инфекцию в зараженном организме.
Диагностика TTV-инфекции базируется на выявлении в сыворотке/плазме вирусной ДНК при помощи ПЦР. Из-за генетической вариабельности вируса большое влияние на чувствительность метода оказывает выбор фрагмента TTV-ДНК, предназначенного для амплификации. Этим, в частности, объясняется разброс результатов по изучению распространения вируса, его источников, путей передачи, клинических экстраполяций — словом, всего, что связано с эпидемиологией и патогенетической значимостью TTV. В последних исследованиях предпочтение отдается нетранслируемому участку генома, который, отличаясь консервативностью, логичнее всего подходит для универсализации ПЦР. Это позволило заметно повысить выявляемость TTV в образцах сыворотки, укрепив представление о его широком распространении в общей популяции.
Вирусоносительство в большинстве регионов Земного шара достигает 80%, а в некоторых странах (Япония, Сингапур, Саудовская Аравия, Мьянмар/Бирма) встречается почти поголовно. Нет необходимости убеждать в высокой контагиозности TTV, хотя механизмы и пути его распространения гипотетичны. Одним из источников является кровь. Поэтому все, что способствует контакту с кровью вирусоносителей, содействует инфицированию. Вместе с тем очевидные и скрытые контакты с зараженной кровью не исчерпывают проблемы — слишком велика инфицированность общей популяции. Более важным (точнее, распространенным) механизмом может быть фекально-оральная контаминация. Об этом говорит присутствие вируса в фекалиях людей с TTV-вирусемией. Высокая устойчивость может поддерживать контаминирующую дозировку даже при небольшом выделении TTV во внешнюю среду.
Высокая степень TTV-инфицированности уже в первые месяцы жизни позволяет думать о вертикальном механизме передачи. TTV-ДНК обнаружена более чем в половине образцов пуповинной крови; ее уровень соответствовал содержанию в материнской крови, склоняя в пользу трансплацентарной передачи. Это вовсе не исключает заражения в раннем постнатальном периоде (например, через грудное молоко, слюну, назофарингеальные секреты), тем более, что не всем авторам удавалось обнаружить TTV-ДНК в пуповинной крови новорожденных от инфицированных матерей.
Участие полового пути передачи сомнительно: существенного повышения TTV-инфицированности и генотипических параллелей в группах риска по заболеваниям, передаваемым половым путем, не установлено. Роль бытовых контактов тоже неубедительна, хотя присутствие вируса в слюне, носоглоточных секретах, коже и волосах допускает такую вероятность. Требует изучения и эпидемиологическая значимость TTV- и TTV-подобных вирусов, циркулирующих среди кур, свиней, коров и овец. Возможно, они не столько расширяют и без того огромный (человеческий) резервуар TTV, сколько содействуют эволюции вируса, создавая дополнительную площадку для образования рекомбинантных клонов. Так или иначе, но эпидемиологической основой TTV-инфекции служит активная персистенция, которая обеспечивает постоянное присутствие инфекционного вируса в крови и (обычно через вирусемию) в других субстратах.
Хроническая вирусемия отражает перманентное размножение TTV в организме. О тканях и типах клеток, где вирус проходит первичную репликацию, а затем персистирует, поддерживая вирусемию, можно сказать следующее. По аналогии с другими однонитевыми ДНК-вирусами следует ожидать, что главным источником репликативного TTV должны быть активно пролиферирующие клетки. Наиболее вероятным резервуаром являются гепатоциты. По крайней мере содержание вируса в биоптатах печени и желчи в 10—100 раз выше, чем в плазме и фекалиях.
Другим источником вируса могут быть лимфоидная ткань и костный мозг, точнее их митотически активные клетки. Если покоящиеся мононуклеары крови включают небольшое количество TTV-ДНК, то при стимуляции фитогемагглютинином они поддерживают репликацию и выделяют значительные количества TTV. Подобно гепатоцитам, клетки костного мозга содержат репликативные (двунитевые) формы TTV-ДНК. После гемотрансфузии TTV-ДНК появляется в крови через одну или несколько недель. Нередко вирус исчезает из циркуляции через несколько недель или лет, но известно и многолетнее (более 20 лет) носительство, позволяющее думать о пожизненной персистенции. Впрочем, и то и другое допускает альтернативное толкование: элиминация из крови не гарантирует удаления вируса из организма, а длительная вирусемия может быть (хотя и нечасто) результатом повторного заражения.
О факторах, содействующих стабилизации TTV-инфекции, можно рассуждать, пользуясь лишь общими представлениями о механизмах вирусной персистенции (см. «Болезнетворность вирусов»). Напомним, в частности, о высокой изменчивости вируса, которая по логике должна способствовать его ускользанию от эффекторов иммунитета. Зависимость от пролиферативной активности клеток может быть причиной того, что в организме в каждую единицу времени репликацию вируса поддерживает небольшая фракция потенциально чувствительных клеток, тогда как остальные (покоящиеся) клетки пребывают в состоянии непермиссивности, давая убежище латентному вирусу. Замечено, что генотипы плазменного TTV и TTV, ассоциированного с мононуклерами крови, не всегда совпадают. Это позволяет думать о компартментализации инфекции, известной для вирусов иммунодефицита человека и гепатита С. Механизмы TTV-вирогении (эписома или коинтеграт с клеточной ДНК) не известны. Попытки обнаружить интегративную форму TTV-генома в гепатоцеллюлярных карциномах и в клетках костного мозга больных лейкозами не увенчались успехом.
Интерес к TTV инициирован декларацией о его причастности к развитию гепатита. Однако взвешенный анализ скорее исключает такую возможность. Негативная позиция (это справедливо и для других претендентов на избирательную гепатотропность) базируется на следующих фактах:
- одинаковое распространение TTV-вирусемии среди больных криптогенным (ни А—Е) гепатитом и у лиц с иной патологией печени или без нее;
- одинаковый уровень сывороточных трансаминаз у лиц с позитивными и негативными показателями TTV-вирусемии;
- отсутствие корреляции между заражением TTV и развитием гепатита, а также между динамикой сывороточных трансаминаз и TTV-ДНК после гемотрансфузий;
- несоответствие между заражением TTV и степенью поражения печени у большинства больных гепатитами В и С;
- отсутствие корреляции между эффективностью лечения хронического гепатита В и С альфа-интерфероном (в сочетании с рибавирином или без него) и элиминацией TTV;
- отсутствие биохимических и гистологических признаков поражения печени у шимпанзе, естественно или искусственно зараженных TTV или TTV-подобными вирусами.
Но возможна и альтернатива. Нельзя, например, исключить участия TTV в этиологии преходящих нарушений печени, регистрируемых по уровню сывороточных трансаминаз. В ряде исследований приводятся данные об утяжеляющем действии TTV на течение гепатита С и фульминантного гепатита. Опираясь на подобные наблюдения и памятуя о способности TTV к репликации в гепатоцитах, можно допустить, что TTV, по крайней мере изредка, причастен к патологии печени. Но если это и так, то речь идет не о стратегии истинно гепатотропных (А—Е) вирусов (для них гепатоциты — единственная или главная патогенетически значимая мишень), а об осложнениях второго (или более глубокого) плана, которые возникают у небольшого числа инфицированных людей и не влекут за собой серьезных последствий. Нечто подобное известно для многих вирусов, лишенных направленной гепатотропности (вирусы кори, краснухи, энтеровирусы, аденовирусы, цитомегаловирус, вирус Эпстайна—Барр, энтеровирусы и др.). Следует помнить о возможной активации TTV другими вирусами, а также о масштабе вирусной нагрузки, т.е. о репликативной активности TTV.
Все сказанное дает основание рассуждать о TTV как о представителе нормальной микрофлоры человека, решившись, наконец, на позитивное переосмысление концепции о вирусах-комменсалах. Впрочем, о TTV продолжают говорить как о вирусе-сироте, которому еще не найдено места в патологии. В этой связи проводятся параллели с другими вирусами, патогенетическое признание которых состоялось спустя годы — вирусы ECHO, реовирусы (оба названия включает акроним «о» от англ. orphan — сирота), аденовирусы, парвовирус В19, вирус Эпстайна—Барр. Вспоминают о цирковирусах животных, способных наносить серьезный ущерб своим хозяевам. Генетическая неоднородность вируса, который может быть совокупностью близко родственных, но разных видов, создает почву для рассуждений о его патогенетической гетерогенности.
Все это интригует, но пока больше выглядит как стремление апологетов TTV выдать желаемое за действительное. Главное, что не подлежит сомнению — это открытие нового вируса с уникальными свойствами, в том числе с необычными взаимоотношениями с человеком. Это, несомненно, будет поддерживать интерес к TTV в ближайшие годы.
Вирусы SEN
Стремление установить природу этиологически неизвестных случаев ни А—Е-гепатита привело к открытию еще одной группы вирусов. Они получили предварительное название SEN-V, по имени больного, послужившего источником выделения. Это одноцепочечный безоболочечный ДНК-вирус, близкий к TTV, содержит около 3900 нуклеотидов, которые включают не менее 3 открытых рамок считывания; отличается генетической неоднородностью. Известно восемь генотипов (A—H), которые различаются примерно на 30% по последовательности ДНК; у человека встречаются два основных варианта — SEN-V-D и SEN-V-H.
Название подчеркивает осторожность нынешних подходов к утверждению значимости новых вирусов в развитии гепатитов. Оказалось, что подобно TTV и HGV SEN-вирус может долго и без каких-либо очевидных последствий находиться в организме человека. Связать его персистенцию с острыми или хроническими заболеваниями печени не удалось, хотя есть данные о том, что он чаще выделяется от больных с посттрансфузионной печеночной патологией. Впрочем, и здесь все не ясно: некоторые авторы склоняются к возможности заражения SEN-V непарентеральным путем. Общий вывод сводится к тому, что SEN-V не играет роли в патогенезе заболеваний печени и может бессимптомно присутствовать у здоровых доноров и больных с непеченочной патологией.
Герпесвирусы
Не бойся собаки брехливой, а бойся молчаливой.
- Персистенция как патогенетическая стратегия.
- Классические и новые герпесвирусы человека.
- Первичные и оппортунистические инфекции.
- Внутриутробные и перинатальные инфекции.
- Онкогенность.
- Проблемы вакцинации и этиотропной терапии.
Семейство Herpetoviridae включает более 80 вирусов человека и животных — от устриц до обезьян. Многие из них сохраняют верность своим хозяевам, избирательно инфицируя определенные виды животных. Во всяком случае, за исключением обезьяньего герпесвируса В, герпесвирусы животных не опасны для людей, что обеспечивает антропонозный характер герпетической патологии человека.
Название семейства происходит от греческого слова herpein — ползти, расползаться. Дело в том, что для герпетических поражений слизистых оболочек и кожи характерны везикулезные высыпания, которые лопаются с образованием расползающихся эрозий. Именно из очага лабиального герпеса (губной лихорадки) был выделен первый вирус этого семейства — вирус простого герпеса.
Герпесвирусы имеют ряд общих признаков, главными из которых являются строение вириона, структура геномной молекулы (ДНК) и ее стратегия в зараженных клетках. Вирион герпесвирусов представляет собой довольно крупные (для вирусов) частицы (150—250 нм в диаметре), которые состоят из нуклеокапсида (кор) кубической симметрии и наружной оболочки (суперкапсид) (рис. 1).

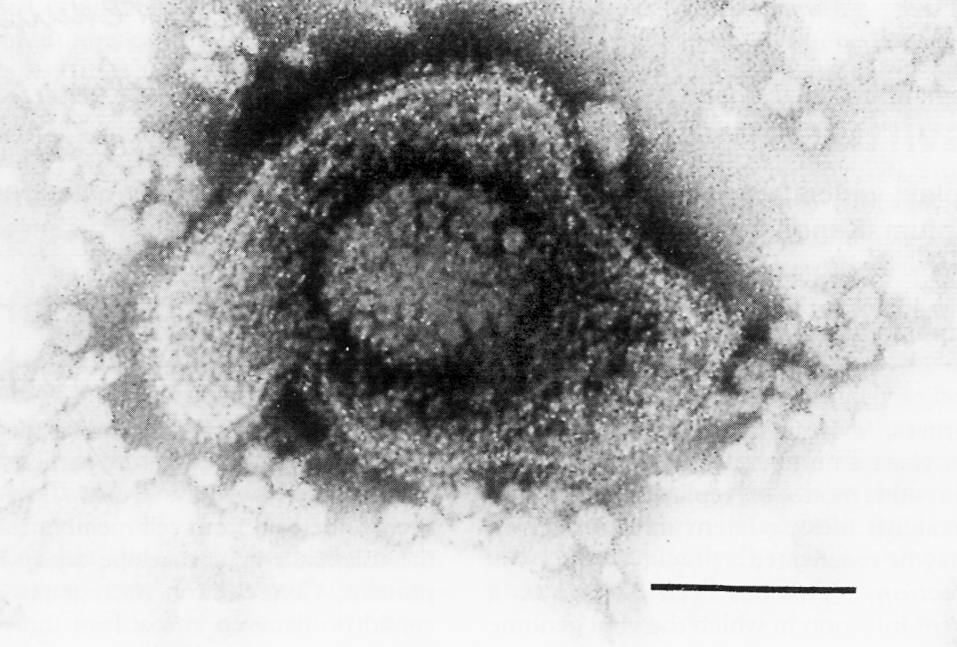
Между ними находится аморфный слой — тегамент (англ. оболочка, покров), содержащий белки, необходимые для инициации репликативного процесса. Нуклеокапсид имеет около 100 нм в диаметре и, как любят подчеркивать вирусологи, построен из 162 капсомеров. Репликация происходит в ядре, здесь же собираются нуклеокапсиды. Суперкапсид образуется из модифицированных вирусом фрагментов ядерной и цитоплазматической мембран. Герпесвирусы практически не отличимы друг от друга под электронным микроскопом, но их легко разграничить по антигенным особенностям вирионных белков и степени гомологии ДНК. Согласно этим критериям, родственные связи между большинством герпесвирусов выражены слабо.
Геном представлен двухспиральной линейной молекулой ДНК. Она устроена своеобразно и кроме уникальных последовательностей включает прямые и инвертированные повторы, позиции которых меняются при репликации вируса (ДНК-изомерия). Значение такого рода перетасовок не известно, но они, безусловно, способствуют мутационным изменениям и в целом — эволюции генома. По крайней мере, согласно рестрикционному анализу (сравнение фрагментов, полученных при расщеплении ДНК специфическими эндонуклеазами — рестриктазами), штаммы, выделенные из разных источников (например, от людей, не имевших между собой контактов), различаются по нуклеотидному профилю ДНК. Необычность герпесвирусной ДНК исключает возможность ее репликации клеточной ДНК-полимеразой: для этого вирусы пользуются собственным ферментом.
Генетическая емкость ДНК позволяет синтезировать 70—100 белков, из которых не менее половины относятся к неструктурным (участвуют в репродукции вируса), а остальные включаются в состав вириона (структурные белки). Широкий набор вирусспецифеских ферментов определяет относительную независимость герпесвирусов от клетки-хозяина, создавая возможность для антивирусных воздействий. Герпетические инфекции (особенно простой герпес) лучше других поддаются химиотерапии, основанной на подавлении уникальных герпесвирусных ферментов — ДНК-полимеразы (ацикловир, ганцикловир, фоскарнет) и ферментов, обеспечивающих кэппинг вирусной мРНК (рибавирин). Новые надежды возлагают на инактивацию вирусспецифической тирозин-протеинкиназы и антисмысловые нуклеотиды.
Репликация герпесвирусов представляет собой хорошо упорядоченный процесс и напоминает воспроизведение других крупных ДНК-вирусов. Литический (продуктивный) цикл начинается с прикрепления вирионов к чувствительным клеткам благодаря высокоизбирательному взаимодействию клеточных рецепторов и суперкапсидных гликопротеинов. Кор освобождается от суперкапсида и транспортируется в ядро, где происходит реализация генетической программы вируса. Последовательность событий включает транскрипцию и трансляцию сверхранних, ранних и поздних генов. Экспрессия пяти сверхранних генов, запускаемая белками тегамента, инициирует активацию дюжины ранних генов, продукты которых необходимы для репликации вирусной ДНК. Вслед за этим в работу включаются остальные (поздние) гены, кодирующие белки вириона. Нуклеокапсидные частицы собираются в ядре и, почкуясь через участки ядерной и плазматической мембран, в которые предварительно включаются и гликозилируются вирусные белки, обретают суперкапсид. Часть зрелых вирионов высвобождается из клеток, но многие, не завершая почкования, связываются с соседними клетками, возбуждая очередной репликативный цикл. Внутриядерная локализация виропласта (место, где образуются нуклеокапсиды) и вирусиндуцированное слияние контактирующих клеток определяют характерную картину цитопатического эффекта — формирование многоядерных клеток (симпласт) с внутриядерными ацидофильными включениями (рис. 2). Нежизнеспособность симпласта ведет к множественной гибели втягиваемых в его построение клеток. Патогенетически важно и то, что внутри симпласта вирус не доступен для антител.


Известно восемь герпесвирусов человека, которые по биологическим особенностям разделены на три подсемейства — альфа, бета и гамма (см. таблицу).
| Подсе-мейство | Цикл роста | Цито- патичность | Персис-тенция | Род | Официальное название | Общеупотре-бительное название |
| Αльфа | Короткий | Цитолитический эффект | Нейроны | Simplexvirus | Герпесвирус человека 1 Герпесвирус человека 2 | Вирус простого герпеса, тип 1 Вирус простого герпеса, тип 2 |
| Varicello-virus | Герпесвирус человека 3 | Вирус ветряной оспы/опоясывающего герпеса | ||||
| Бета | Длинный | Цитомегалический эффект | Железы, почки | Cytomegalovirus | Герпесвирус человека 5 | Цитомегаловирус |
| Лимфопроли-феративный эффект | Лимфоидная ткань | Roseolovirus | Герпесвирус человека 6 Герпесвирус человека 7 | Герпесвирус человека, тип 6 Герпесвирус человека, тип 7 | ||
| Гамма | Вариабельный | Лимфопроли-феративный эффект | Лимфоидная ткань | Lympho-cryptovirus | Герпесвирус человека 4 | Вирус Эпстайна—Барр |
| Rhadinovirus | Герпесвирус человека 8 | Герпесвирус человека, тип 8 |
Альфа-вирусы прекрасно размножаются в эпителиальных клетках и фибробластах человека и животных. Цитомегаловирус (бета) медленно реплицируется в фибробластах человека, неохотно выделясь из них. К бета-вирусам отнесены также герпесвирусы 6 и 7, хотя их особенностью является поражение Т-лимфоцитов, и по этому признаку они лучше подходят к лимфотропным гамма-вирусам. Гамма-вирусы культивируются в В- (вирус Эпстайна—Барр) и Т-лимфоцитах (герпесвирус 8). Они не убивают, а вынуждают размножаться свои мишени, реально претендуя на участие в развитии опухолей, прежде всего лимфопролиферативных заболеваний. Острая деструктивная патология наиболее характерна для альфа-вирусов.
Несомненно, герпетическая инфекция — самая распространенная вирусная инфекция человека. Из-за неустойчивости во внешней среде заражение происходит при тесных контактах, допускающих прямую передачу вируса новому хозяину. Первичная контаминация часто остается бессимптомной, хотя почти поголовное инфицирование делает ее весьма ощутимой в абсолютном исчислении. Проявления герпетической инфекции многолики — от безобидной губной лихорадки до генерализованных форм, врожденных уродств и, возможно, злокачественных опухолей; от спорадических заболеваний до эпидемических вспышек. Болезнетворность во многом связана с длительной, часто пожизненной персистенцией герпесвирусов в организме естественных хозяев, создающей постоянную угрозу реактивации. Формула «однажды инфицирован — инфицирован на всю жизнь» отлично подходит к герпесвирусам. Получив их в детстве или даже в утробе матери, едва ли не каждый из нас остается носителем на всю жизнь.
Для закрепления в организме герпесвирусы пользуются излюбленными типами клеток, сохраняясь в них в виде копий кольцевидных ДНК автономно от хромосом (неинтегративная вирогения). В латентной фазе экспрессируется ограниченное число вирусных генов, продукты которых поддерживают вирус в неактивном состоянии. Реактивация, т.е. переход от персистенции к полномасштабной репликации — едва ли не главное содержание герпетических инфекций, которое отражает срыв контролирующих (сдерживающих) механизмов хозяина. В целом о взаимоотношениях с герпесвирусами следует судить не только по клиническим, но и по вирусологическим (элиминация или персистенция вируса) исходам инфекции, учитывая патогенетические ресурсы эндогенных вирусов (рис. 3). Следует помнить, что антивирусные препараты не оказывают влияния на персистенцию герпесвирусов.


При всех герпетических инфекциях в крови появляются антивирусные антитела. Нередко это беспомощные свидетели, которые не обеспечивают выздоровления и не предупреждают рецидивов. В лучшем случае они отражают устойчивость к вирусемии, патогенетически облигатной для единственной герпетической инфекции — ветряной оспы. Главная роль в защите от герпесвирусов принадлежит цитотоксическим Т-лимфоцитам, атакующим зараженные клетки. Впрочем, и они не гарантируют от реактивации эндогенных вирусов, а иногда даже участвуют в повреждении тканей.
Специфическая иммунопрофилактика (живая аттенуированная вакцина) разработана только для ветряной оспы.
Вирус простого герпеса
Эпитет «простой» не следует понимать буквально. Он не отражает сложного устройства вируса, а тем более многоликости его взаимоотношений с человеком. Начнем с того, что имеются две разновидности вируса простого герпеса — ВПГ-1 и ВПГ-2. Обладая генетическим родством (примерно 50% гомологии ДНК), они различаются по экологии, патогенетическим потенциям и антигенным свойствам.
ВПГ-1 обычно проникает через эпителий слизистой оболочки ротовой полости, рото- и носоглотки (рис. 4), хотя входными воротами могут быть и другие участки тела. Первичная контаминация обычно происходит в раннем детстве при контакте с инфицированной слюной (в первые 6 мес новорожденные защищены материнскими антителами). Но заразиться можно и позже, о чем говорит прослойка серонегативных (лишенных анти-ВПГ-1-антител) детей и взрослых, соответственно 60—80 и 30—50%. Вирус обладает слабой вирулентностью, и лишь изредка заражение проявляется клинически. У детей это хорошо известная молочница — афтозный (везикулезно-эрозивный) гингивостоматит, который протекает доброкачественно, хотя и не заканчивается вирусологическим выздоровлением, т.е. элиминацией вируса. Впрочем, и при бессимптомной инфекции вирус не покидает организм.


Через нервные окончания он проникает в регионарные ганглии чувствительных нервов, где сохраняется в ядрах нейронов в виде ДНК. Для ротовой полости регионарными служат ганглии тройничного нерва, и именно здесь ВПГ-1 находит постоянное убежище. Отсюда он периодически эмигрирует на периферию, бессимптомно выделяясь в слюну либо повреждая (реактивация!) эпителиоциты в зоне, обслуживаемой зараженными нейронами (рис. 5). Нейротропность ВПГ-1 проявляется и в том, что после реактивации в тройничном ганглии он может пойти по восходящему пути (в головной мозг) с развитием смертельно опасного энцефалита. К счастью, это бывает редко, но в развитых странах служит едва ли не главной причиной такого рода патологии.


Латенция и реактивация вируса — единый с биологической точки зрения процесс, последствия которого определяются числом вирусных частиц, реплицируемых в эпителиальных клетках. В нейронах вирус не размножается, так как транскрипция его генов здесь заблокирована. Экспрессируется лишь несколько генов, продукты которых сдерживают активность вирусной ДНК. К стимулам, которые способствуют оживлению вируса, относятся солнечный загар, инфекции, ослабление иммунитета, эмоциональный стресс, менструация. Клинические проявления реактивации ВПГ-1 (т.е. вторичной инфекции) включают рецидивы лабиального герпеса (губной лихорадки), кожного герпеса (особенно лица), кератоконъюнктивита (офтальмогерпеса). Патогенез наиболее травматичных вариантов офтальмогерпеса (главной причины слепоты в развитых странах) включает иммунологические механизмы — замедленную гиперчувствительность к вирусным антигенам, аутоагрессивные реакции Т-лимфоцитов, иммунокомплексное воспаление.
В типичных случаях возникают высыпания из нескольких сгруппированных пузырьков (везикул) размером 1,5—2 мм на фоне эритемы и отека (рис. 6). Этому предшествуют неврологические симптомы в виде жжения, покалывания, зуда, реже боли — результат прямого вирусного поражения нервов и/или воспалительной реакции вокруг нервной ткани. Везикулы лопаются, образуя болезненные эрозии (язвы), которые бесследно заживают, подвергаясь эпителизации. Нередко высыпания рецидивируют на одном и том же месте. Это отражает регионарность иннервации и привязку вирусной персистенции к определенным нейронам. Процесс, не осложненный вторичной инфекцией, разрешается через 10—14 (по другим данным, через 5—7) дней.

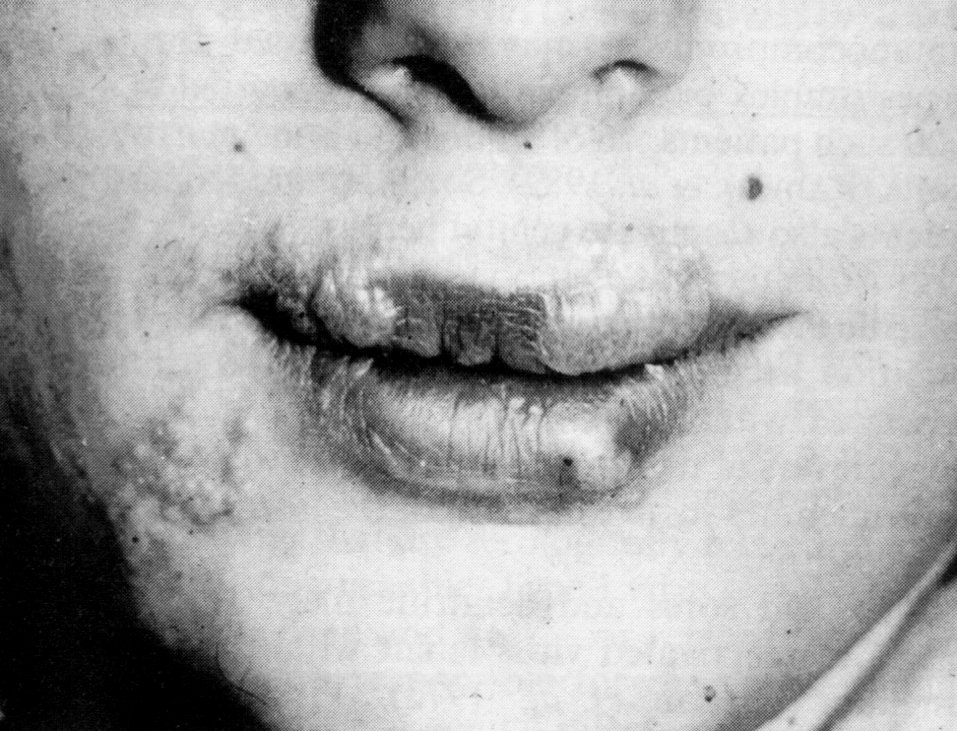
ВПГ-2 отличается повышенным тропизмом к эпителию генитальной сферы (рис. 7). Первичное заражение происходит по достижении половой зрелости, при половых контактах. Это отражает и процент серопозитивности среди детей и взрослых — 0—5 и 20—50%. Подобно ВПГ-1, вирус обычно не вызывает клинически значимых поражений, но закрепляется в организме, транспортируя свою ДНК в регионарные (сакральные) ганглии задних корешков спинного мозга. Лишь у 1—15% инфицированных через 2—3 дня появляются везикулезно-эрозивные высыпания на эритематозно-отечной слизистой оболочке и коже полового члена, вульвы, влагалища, шейки матки, промежности. Они болезненны, сопровождаются жжением и саднением, а иногда упорной невралгией. Значительное количество вируса в везикулезной жидкости (1—10 млн. в 1 мл) определяет высокую контагиозность больного. В сочетании со склонностью к рецидивам (они наблюдаются у 50—75% больных, продолжаются 2—3 нед и могут повторяться 7—20 раз в год) это делает генитальный герпес одним из самых распространенных заболеваний, передаваемых половым путем. Широкому распространению инфекции способствует бессимптомное течение цервикального герпеса у 40—75% женщин.
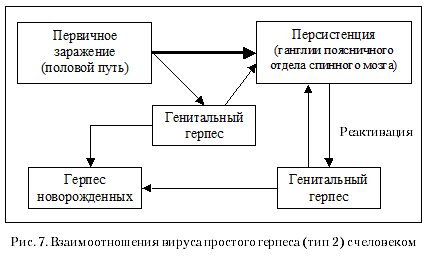

Широко обсуждалась связь между ВПГ-2 и раком шейки матки. К этому побуждали данные о выделении вируса из опухолевой ткани, повышенные титры антигерпетических антител у больных женщин и трансформирующие потенции ВПГ-2 в эпителиальных культурах. Сегодня прямое участие ВПГ-2 в происхождении рака отрицается. Главную роль играют папилломавирусы (см. «Папилломавирусы»).
Говоря о своеобразии взаимоотношений ВПГ-1 и ВПГ-2 с человеком, т.е. о преимущественном инфицировании каждым из них определенных участков тела («выше и ниже пояса»), следует помнить об известной условности такого деления, ставшей особенно заметной за три последних десятилетия (возможно, в связи с расширением практики орогенитальных половых контактов). При сохранении общей тенденции ВПГ-1 нередко инфицирует половую сферу (10—30% от общего числа больных генитальным герпесом), а ВПГ-2 выявляется у 10—20% больных с экстрагенитальными герпетическими поражениями.
Диагноз простого герпеса не представляет трудностей. Цитологическим подтверждением служит присутствие в соскобах из пораженных участков многоядерных клеток с внутриядерными ацидофильными включениями (см. рис. 2). Для большей убедительности препараты можно обработать мечеными антигерпетическими антителами (иммунофлюоресцентная микроскопия). Созданы коммерческие тест-системы для обнаружения ВПГ-антигенов (иммуноферментный анализ) и ДНК (полимеразная цепная реакция). Благодаря непривередливости и быстрому размножению в клеточных культурах ВПГ можно выделить из очагов поражения. Это рекомендуется делать у беременных женщин для снятия угрозы неонатального герпеса.
Определение противогерпетических антител (нарастание титра, выявление IgM-антител) имеет значение лишь при первичной инфекции, так как в дальнейшем они обнаруживаются у большинства людей, а рецидивы, как правило, не дают серологических сдвигов. Не ясна и протективность антител. В культурах in vitro антитела против оболочечных ВПГ-антигенов ограничивают заражение соседних клеток, а при добавлении комплемента инактивируют вирус. Однако в герпетических высыпаниях ВПГ-1 и ВПГ-2 сохраняют инфекциозность несмотря на присутствие антител. Главная роль принадлежит цитотоксическим Т-лимфоцитам. Об этом говорит склонность к генерализации герпеса у больных с дефектами Т-клеточного иммунитета при нормальной резистентности на фоне недостатка иммуноглобулинов.
Было немало попыток создать вакцину против ВПГ, в основном против редицивов офтальмо- и генитального герпеса. К сожалению, все они не позволяют избавиться от вируса, сохраняя эпидемиологически опасное выделение ВПГ.
Вирус ветряной оспы/ опоясывающего герпеса
Мысль о том, что ветряную оспу (varicella — лат. пятнистый) и опоясывающий герпес (herpes zoster — греч. пояс) вызывает один и тот же агент, возникла в конце ХIХ в. из наблюдений о заражении детей ветряной оспой при контакте с больными опоясывающим лишаем (устаревший синоним опоясывающего герпеса). Прямые доказательства были получены позже в опытах с заражением добровольцев материалом из кожных высыпаний (везикул) больных зостером: у некоторых из них возникала ветряная оспа. Труднее утверждалось представление об эндогенной природе опоясывающего герпеса, т.е. о реактивации эндогенного вируса, сохранившегося в организме после перенесенной ветряной оспы. До сих пор появляются сообщения о случаях зостера, возникших, якобы, после контакта с больными опоясывающим герпесом или ветряной оспой, т.е. в результате экзогенного заражения. Но большинство убеждено в противном, признавая за зостером эндогенное происхождение.
В отличие от вируса простого герпеса и других герпесвирусов, первичное инфицирование вирусом ветряной оспы/опоясывающего герпеса (varicella-zoster virus, V-Z-вирус) редко остается бессимптомным (рис. 8). Как правило, через 10—20 дней (в среднем через 2 нед) развивается характерный клинический синдром — ветряная оспа. Обычно болеют дети (чаще 5—9 лет), и мало кому удается избежать заражения и не заболеть в детском возрасте. В большинстве случаев заболевание протекает легко, без интоксикации и осложнений. Варицелла взрослых повторяет клинику детской ветряной оспы, но протекает тяжелее и чаще дает осложнения.
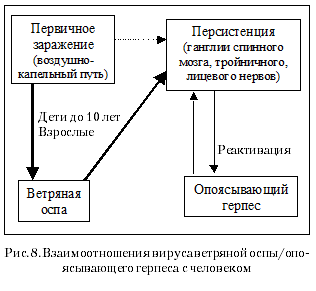


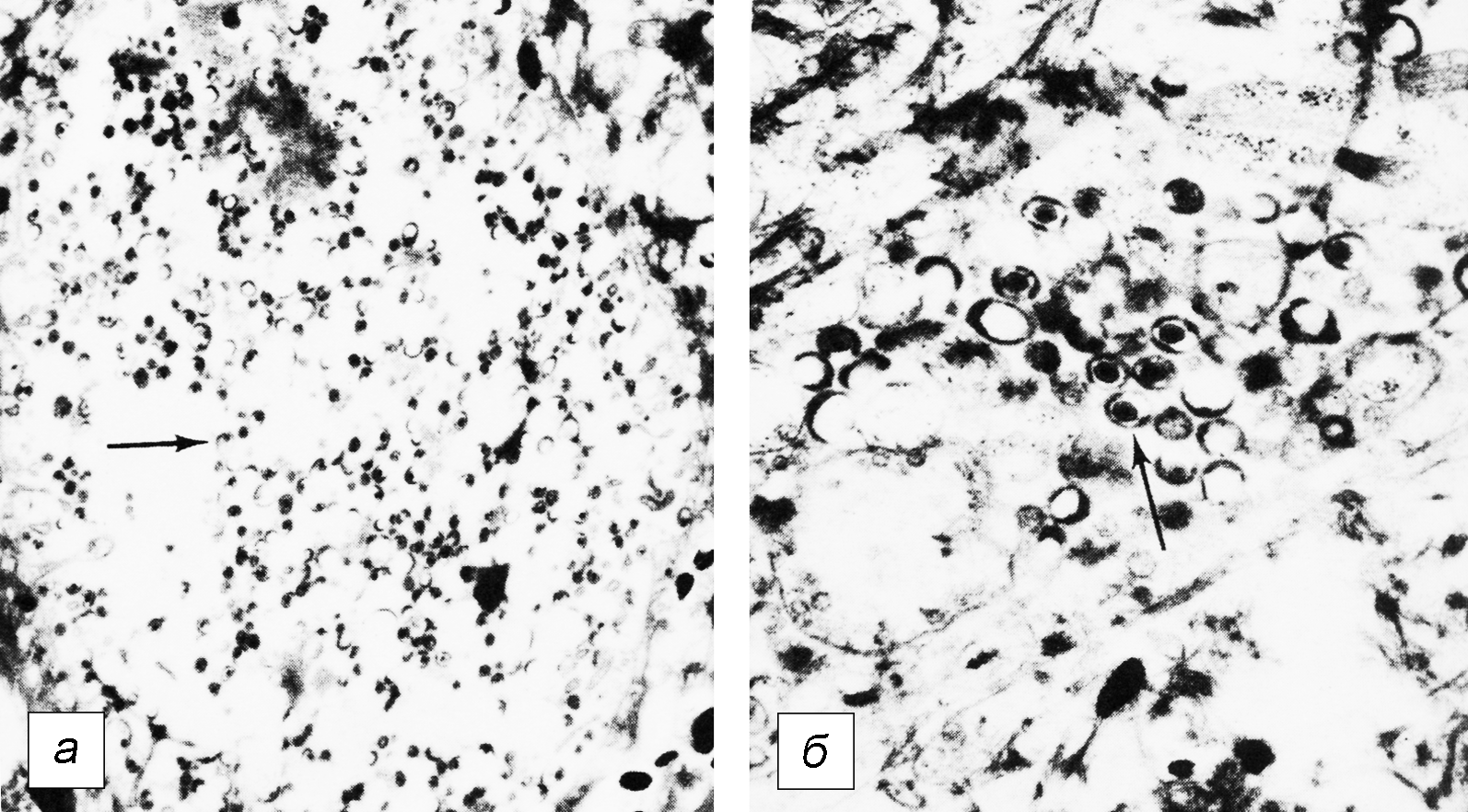
Обычно они эволюционируют по схеме «макула — папула — везикула — вскрытие (или резорбция везикулы) — образование корочки — бесследное заживление». Превращение макул (красноватых пятен диаметром 2—4 мм) в везикулы происходит очень быстро, в течение нескольких часов. Общая продолжительность периода высыпаний (от появления первых элементов до образования корочек на месте лопнувших везикул) составляет 2—3 дня. Если учесть, что вслед за первыми появляются новые очаги сыпи, которые подвергаются аналогичной трансформации, то пятнисто-папулезно-везикулезную сыпь можно видеть на протяжении 2—10 дней. Корочки отпадают через 5—10 дней после образования. Большинство высыпаний локализуется на коже туловища, хотя могут поражаться и другие участки тела — лицо, шея, конечности. Иногда поражаются слизистые оболочки полости рта и половых органов (особенно у девочек) — энантема (т.е. «внутренняя сыпь»). В редких случаях развивается афтозный стоматит, похожий на гингивостоматит при простом герпесе. Наиболее вероятное осложнение — вторичная пиогенная инфекция содержимого везикул. У 15% взрослых поражаются легкие (пневмония), но процесс обычно завершается самоизлечением.
Известна возможность трансплацентарного заражения плода от женщин, заболевших ветряной оспой во время беременности. Обычно это не имеет серьезных последствий, так как антитела, продуцируемые матерью, успевают защитить плод от вирусемии (по той же причине дети до года редко болеют ветряной оспой). Но если с момента высыпаний до родов проходит менее 5 дней, то из-за отсутствия у матери антител (первичная инфекция!) плод не получает пассивной защиты и родившийся ребенок может заболеть тяжелой ветряной оспой. Это связано с тем, что при трансплацентарной передаче вирус внедряется не через респираторный тракт, а непосредственно в кровь. Если мать заболевает вскоре после родов и заражает новорожденного обычным способом, заболевание протекает доброкачественно. К счастью, все это скорее исключение, чем практическая реальность: переболев ветряной оспой в детстве, почти все женщины серопозитивны и устойчивы к V-Z-вирусу.
После выздоровления вирус не удаляется из организма (см. рис. 8). Подобно ВПГ, он проникает в регионарные ганглии чувствительных нервов и персистирует в нейронах в виде геномной молекулы. Поскольку при ветряной оспе поражаются разные участки тела, вирус получает возможность для распространения в межпозвоночные ганглии всех уровней спинного мозга, а также в ганглии тройничного и лицевого нервов.
Сочетание высокой вирулентности с пожизненной персистенцией возбудителя — парадокс, в который непросто поверить. Но сегодня мало кто сомневается в такой возможности, как и в том, что латенция определяет патогенетическую двуликость V-Z-вируса, т.е. способность вызывать ветряную оспу (первичная экзогенная инфекция) и опоясывающий герпес (реактивация эндогенного вируса). После перенесенной ветряной оспы иммунитет к реинфицированию остается на всю жизнь и скорее всего связан с антителозависимой устойчивостью к вирусемии. Напротив, резистентность к эндогенному вирусу не абсолютна. Обеспечивая устойчивость к рецидивам ветряной оспы (генерализованному процессу), антитела не спасают от местных проявлений реактивации вируса. Повышенная чувствительность к опоясывающему герпесу у пожилых людей и больных с дефектами клеточного иммунитета говорит о том, что сдерживающим началом против эндогенного вируса являются Т-лимфоциты, хотя и они не обеспечивают полной защиты.
Внешним признаком опоясывающего герпеса являются везикулезно-эрозивные высыпания на коже, напоминающие экзантему при ветряной оспе. Они имеют характерную локализацию, возникая по ходу чувствительных нервов. Чаще поражаются зоны, иннервируемые из грудных ганглиев спинного мозга. В таких случаях сыпь опоясывает туловище от позвоночника до средней вентральной линии (рис. 10). Почти столь же типичны поражения лица, особенно по ходу глазного ответвления тройничного нерва (рис. 11); вовлечение в процесс роговицы чревато нарушением зрения. Поражения других участков встречаются реже, причем, как правило, атакуется единственный сегмент тела.

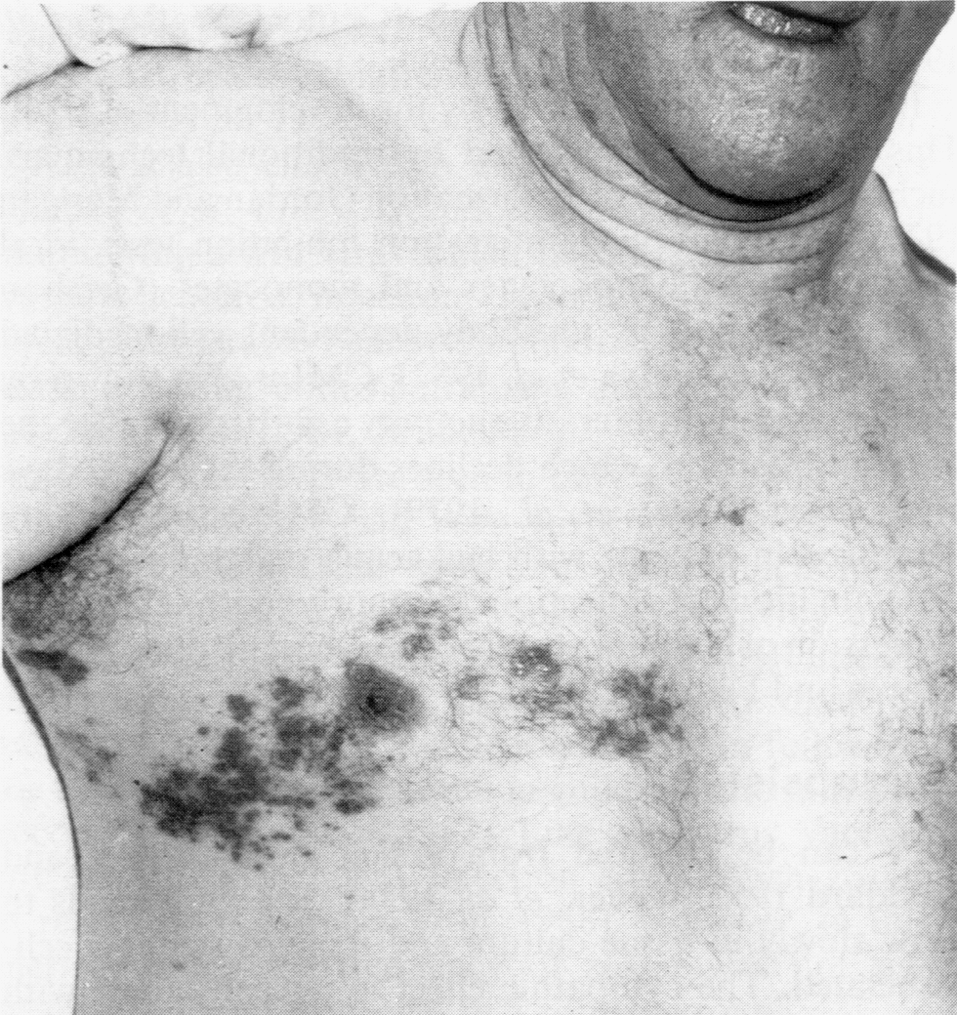

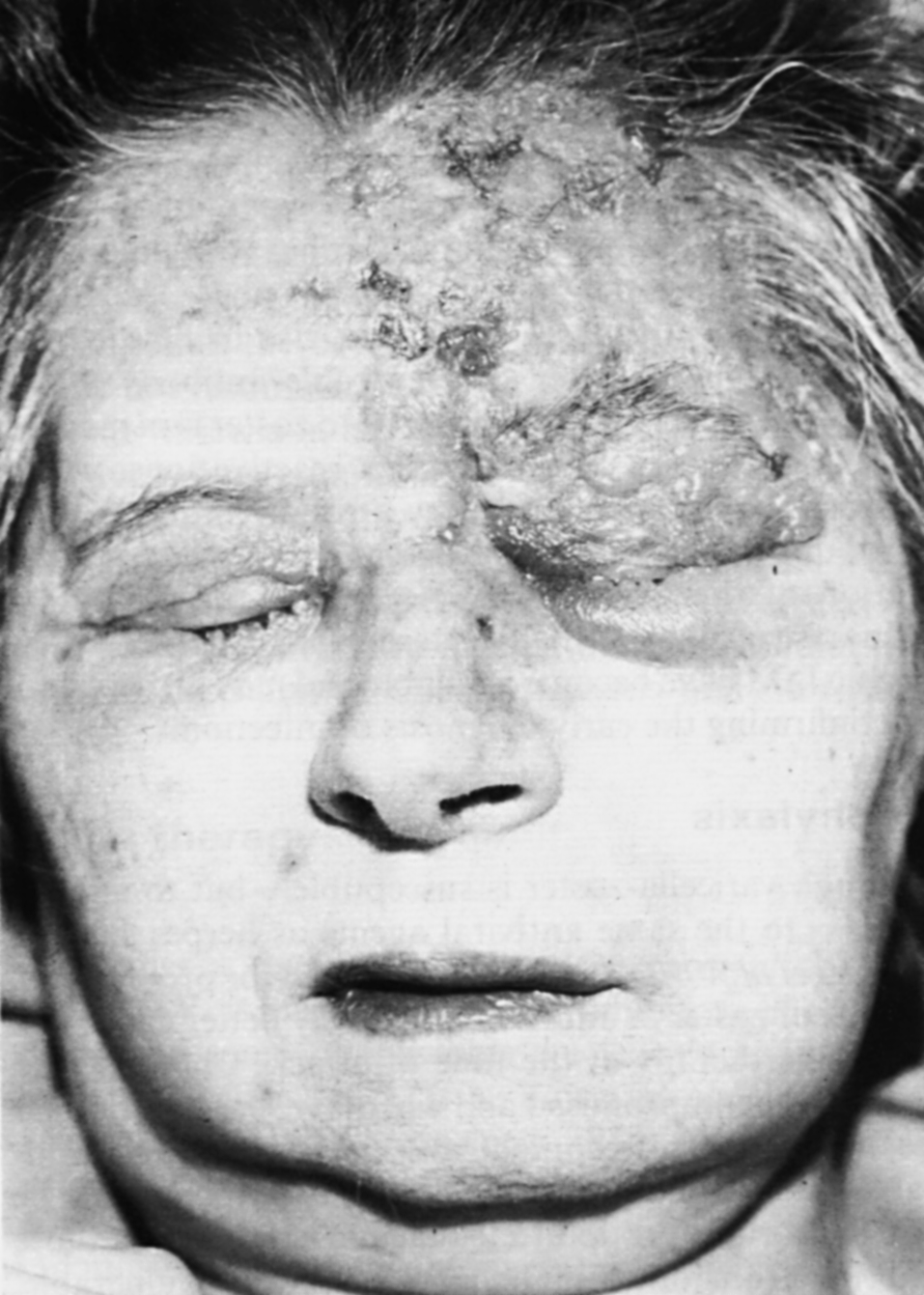
Но первым симптомом, который на несколько дней опережает экзантему, сопутствует ей и сохраняется после ее исчезновения, является резкая боль по ходу нервов поражаемого кожного сегмента. Не исключено, что невралгии (особенно тройничного нерва) возникают и без кожных высыпаний, оставаясь этиологически не расшифрованными. Патогенез болевого синдрома не ясен. Можно думать, что в воспалительный процесс вовлекаются корешковые ганглии и даже задние рога спинного мозга, но нельзя забывать и о раздражении периферических нервных окончаний, тем более что при зостере (в отличие от ветряной оспы и простого герпеса) в везикулах много нейтрофилов, знаменующих обострение процесса.
Обычный исход опоясывающего герпеса — клиническое выздоровление, повторение заболевания является необычным. Процесс опасен лишь для больных с дефектами иммунитета (лимфопролиферативные заболевания, СПИД, химиотерапия по поводу злокачественых опухолей). В подобных случаях возможна генерализация процесса с поражением внутренних органов, нервной системы и высокой летальностью, тем более что эффективность антигерпетических препаратов при данной патологии оставляет желать лучшего. Больные не опасны для лиц, перенесших ветряную оспу, но могут заразить тех, кто еще не сталкивался с вирусом.
Живая аттенуированная вакцина обеспечивает великолепную защиту от ветряной оспы, но не гарантирует ее от опоясывающего герпеса.
Цитомегаловирус
Название цитомегаловируса (ЦМВ) отражает морфологию его цитопатического эффекта. В очагах поражения выявляются клетки больших размеров (25—40 мкм; греч. megas — гигантский) с крупными внутриядерными включениями, отграниченными от ядерной мембраны бледным, не воспринимающим окраску ободком (рис. 12).

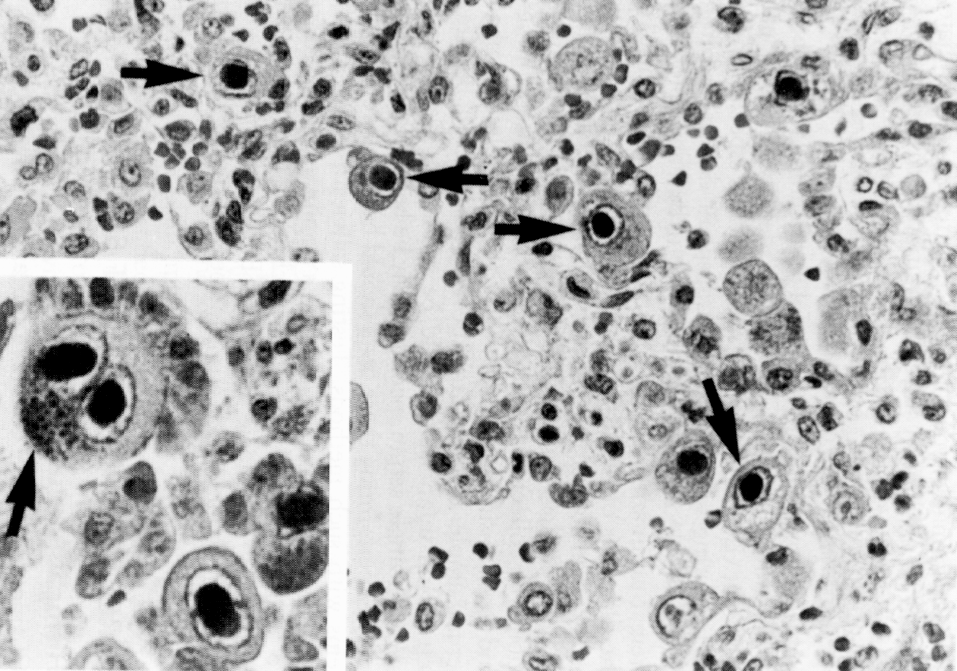
Такого рода клетки (их образно сравнивают с совиным глазом) впервые обнаружены в 1904 г. в почках, легких и печени новорожденных с диагнозом «врожденный сифилис». Позже (1921—1926 гг.) был предложен термин «цитомегалия» и высказано предположение о ее вирусной природе. Так как цитомегалические клетки часто находили в аутопсийных слюнных железах, гипотетический вирус долго фигурировал как «вирус слюнных желез». Современное название появилось в конце 1950-х гг. после того, как ЦМВ научились культивировать в фибробластах человека. Сходные вирусы обнаружены и у животных — обезьян, мышей, морских свинок и др. Человек не чувствителен к чужим ЦМВ, а его собственный вирус (который достаточно неоднороден, но тем не менее создает межштаммовый иммунитет) размножается только в культуре человеческих фибробластов. Удивительно, но эпителиальные клетки, которые поражаются в организме, устойчивы к вирусу in vitro. Впрочем, фибробласты тоже довольно неохотно поддерживают репликацию ЦМВ, и цитопатический эффект в их культурах заметен не ранее 30—50 дней. Вирус прочно связан с клетками и лишь в небольшом количестве сбрасывается в культуральную среду. Поэтому заражение новой порции клеток лучше всего удается путем прямого контакта (сокультивирования) с зараженными клетками.
Считается, что к пятидесятилетнему возрасту инфицируются 75%, а по некоторым данным, почти 100% людей. В отличие от других герпесвирусов, этот показатель повышается медленно, примерно с 1%-ным ежегодным приростом. Гораздо быстрее контаминируются новорожденные: к концу первого года жизни 5—10% детей обретают серопозитивность. Заражение происходит в родовом канале, при грудном вскармливании или через слюну. Но это может случиться и раньше: обладая склонностью к трансплацентарной передаче, ЦМВ инфицирует 0,3—2,4% плодов. В дальнейшем главным источником вируса служит слюна, но так как он содержится и в других биологических жидкостях (моча, цервикальный и вагинальный секреты, сперма, кровь, пищеварительные секреты), пути заражения могут быть разными: воздушно-капельный, половой, парентеральный. С развитием трансплантологии реальным стало заражение ЦМВ при пересадке органов, особенно почек. Это опасно для серонегативных реципиентов, не имевших ранее контактов с ЦМВ.
Подобно всем герпесвирусам, для ЦМВ характерно длительное (часто пожизненное) пребывание в организме (рис. 13). Главным резервуаром служат мононуклеарные фагоциты (моноциты, макрофаги). Вирус содержится и в нейтрофилах, но, учитывая их короткую жизнь, они вряд ли способны поддерживать персистентную инфекцию. Следует скорее задуматься над вероятностью скрытого заражения костно-мозговых предшественников с поэтапным (при миелопоэзе) переходом вируса в зрелые лейкоциты. Наконец, ЦМВ определяется в эпителиоцитах слюнных желез, почечных канальцев, выводных протоков поджелудочной железы, в гепатоцитах и других тканях. Присутствие в них ЦМВ не всегда отвечает критерию инертной персистенции, так как, подвергаясь репликации, вирус не только сбрасывается в окружающую среду, но и вызывает ограниченные поражения, которые регистрируются по появлению цитомегалических клеток.


Клинически ЦМВ впервые заявил о себе как возбудитель патологии плода и новорожденных. Внутриутробное инфицирование обычно проходит незаметно и ничем не обнаруживает себя при рождении. Но в 5% случаев (чаще при заражении в двух первых триместрах беременности) развивается цитомегалическая болезнь — острая форма инфекции с поражением внутренних органов. Характерны дефекты развития плода, гепатит и гепатоспленомегалия, тромбоцитопеническая пурпура (геморрагический синдром), гидро- и микроцефалия, хориоретинит. Это ведет к снижению интеллекта, локомоторным расстройствам, потере зрения. Прогноз всегда суровый, вплоть до мертворождения. Но и отсутствие клинических симптомов к моменту родов не означает полного подавления внутриутробной инфекции. В 15% случаев процесс получает медленную, но опасную эволюцию. Новорожденные отстают в умственном развитии и теряют слух из-за поражения сенсорных центров слухового нерва.
Необходимым условием для трансплацентарной передачи ЦМВ является вирусемия беременных. Ее причиной может быть реактивация латентного вируса (вторичная инфекция) или заражение женщин, не имевших ранее контакта с ЦМВ (первичная инфекция). Второй вариант более опасен. Дело в том, что если в первом случае плод одновременно с вирусом получает нейтрализующие антитела, то первично инфицируемые женщины серонегативны, а потому не готовы к экстренной защите плода от ЦМВ. Сегодня есть возможности для выявления антител (IgM, IgG) против ЦМВ. Это необходимо, чтобы решить вопрос об иммунологическом статусе беременных женщин и тем самым определить степень риска первичного заражения цитомегаловирусами. Предложены системы для поиска ЦМВ-антигенов и ПЦР с целью выявления ЦМВ-ДНК. Все это снижает опасность инфицирования плода, хотя и не гарантирует полной защиты от заражения — эндогенного или экзогенного.
Механизм, благодаря которому вирус ускользает от антител, циркулируя в крови серопозитивных женщин (т.е. при реактивационной вирусемии), не ясен. Возможно, вирус прячется внутри лейкоцитов (лейковиремия) и/или покрывается белками хозяина (например, бета-2-микроглобулином). Для матери вирусемия не опасна.
Постнатальное заражение почти всегда бессимптомно. Иногда возникает синдром инфекционного мононуклеоза, но без характерных для него гетерофильных антител. Полагают, что ЦМВ отвечает за 20—50% таких заболеваний (они не имеют отношения к ВЭБ-инфекционному мононуклеозу, см. ниже). Изредка на первый план выходят признаки гепатита или пневмонии, которые, впрочем, быстро проходят. Клинически значимой реактивации ЦМВ-инфекции у здоровых людей не бывает.
Ситуация резко меняется при ослаблении Т-клеточного иммунитета. ЦМВ (реактивированный или экзогенный) — один из главных возбудителей оппортунистических инфекций при СПИДе, у реципиентов аллотрансплантатов и других больных, принимающих иммунодепрессанты и цитостатики. Инфекция клинически многолика — от очаговой цитомегалии до генерализованного процесса с поражением внутренних органов и центральной нервной системы. Помочь таким больным трудно, так как ЦМВ-инфекция не поддается или плохо поддается воздействию антигерпетических средств. Не случайно в США появилась информация о попытках применения для лечения ЦМВ-инфекций препарата на основе антисмысловых ЦМВ-нуклеотидов (коммерческое название benzimidavir).
Лечение альфа-интерфероном не имеет успеха. Не следует переоценивать и эффективность гипериммунных анти-ЦМВ-иммуноглобулинов: возлагавшиеся на них надежды скорее не оправдали себя. Впрочем, этого следовало ожидать, исходя из патогенеза цитомегалии. Вирус распространяется при плотных межклеточных контактах, плохо доступных для антител, а пример с клинически значимой (для плода) вирусемией серопозитивных матерей говорит о сохранении болезнетворных потенций ЦМВ в присутствии антител.
Цитотоксические Т-лимфоциты уничтожают ЦМВ-инфицированные клетки, но это имеет и негативные последствия, если учесть, например, что ЦМВ-пневмония может иметь иммунологическую основу. Этим, в частности, объясняется ее резистентность к противовирусным препаратам, приносящим определенную пользу при других вариантах ЦМВ-инфекции, когда клетки повреждаются самим вирусом.
Испытывается несколько вакцин для профилактики ЦМВ-инфекции у серонегативных доноров. Это касается прежде всего беременных женщин и реципиентов аллогенных тканей.
Вирус Эпстайна—Барр
Вирус назван в честь Мишеля Эпстайна и Эвелины Барр, выделивших его в 1964 г. из лимфомы Беркитта. Эта злокачественная, быстро прогрессирующая опухоль стала знаменитой в 1958 г. после того, как английский врач Д. Беркитт обратил внимание на ее эндемичность для Центральной Африки (позже такое же наблюдение сделано для Новой Гвинеи). Лимфома Беркитта (или африканская лимфома) поражает детей 5—8 лет и первично локализуется в зоне верхней челюсти, вызывая ее деструкцию (остеолиз). В процесс вовлекается центральная нервная система, возможны метастазы в другие органы. Несмотря на пугающую картину (рис. 14), химиотерапия обычно быстро ведет к обратному развитию опухоли и рубцеванию пораженной ткани.


Предположение о вирусной природе лимфомы Беркитта (Д. Беркитт считал, что возбудителем является арбовирус, переносимый комарами) инициировало множество исследований по выделению гипотетического возбудителя. Признание получил вирус, обнаруженный М. Эпстайном и Э. Барр — ВЭБ. Репутация его как онкогенного вируса укрепилась после выделения ВЭБ еще из одной злокачественной опухоли — карциномы носоглотки, эндемичной для Юго-Восточной Азии.
Но вскоре отношение к ВЭБ резко изменилось. После случайной внутрилабораторной вспышки инфекционного мононуклеоза (заболело несколько сотрудников одной из лабораторий США, изучавших онкогенность ВЭБ) и анализа распространенности анти-ВЭБ-антител среди здоровых людей миф об эндемичности вируса и его онкогенной облигатности был рассеян.
Оказалось, что ВЭБ встречается повсеместно, причем большинство людей инфицируется в детстве, и к трем годам 30—80% (в экономически отсталых странах почти 100% — сказывается плотность контактов!) становятся носителями ВЭБ. Заражение обычно проходит бессимптомно, но иногда возникает патология — инфекционный мононуклеоз (рис. 15). В юношеском возрасте болеет около половины первично инфицированных (т.е. анти-ВЭБ-серонегативных), после 35—40 лет заболевание почти не встречается.

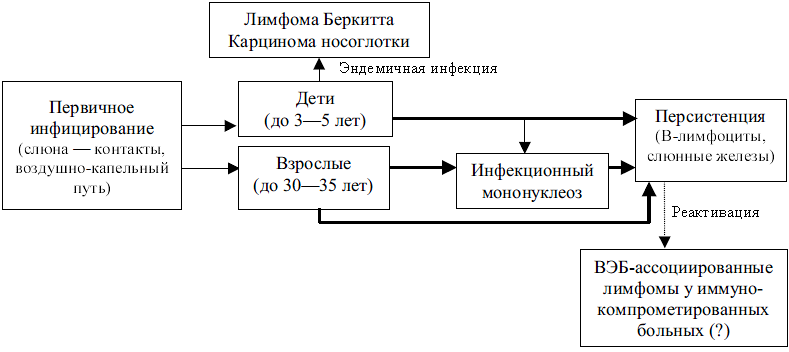
Вирус выделяется в слюну и распространяется с орофарингеальными секретами. Особенно опасны прямые контакты, что оправдывает одно из старых названий инфекционного мононуклеоза — болезнь поцелуев (англ. kissing disease). Инкубационный период составляет 5—12 дней, но нередко затягивается до 4—7 нед. Это то время, которое вирус использует для размножения в эпителиоцитах ротоглотки и околоушных железах, подготавливая заражение В-лимфоцитов, взаимоотношения с которыми служат базой ВЭБ-инфекции.
В-лимфоциты инфицируются при контакте с эпителием носоглотки, где первично размножается вирус. Они связывают ВЭБ при помощи одного из рецепторов для производных С3-фактора комплемента (CD21, т.е. CR2) и сохраняют его внутриклеточно в виде множества внехромосомных копий ДНК. Репликации вируса (образования вирионов) в В-лимфоцитах не происходит. Однако эта латенция относительна. ВЭБ экспрессирует некоторые из своих генов, синтезируя белки, которые появляются в составе HLA на поверхности зараженных клеток и служат мишенью для Т-лимфоцитов (такие же антигены обнаруживаются и на первично инфицированных эпителиоцитах). Кроме того, ВЭБ-инфекция вызывает трансформирующий эффект, повышая способность В-лимфоцитов к размножению. In vitro такие лимфоциты дают начало «бессмертным», активно пролиферирующим линиям клеток, которые формируют «микроопухоли», регрессирующие под влиянием Т-киллеров, полученных от тех же доноров. Сходный механизм действует, по-видимому, и in vivo, защищая хозяина от опухолевой экспансии ВЭБ-трансформированных клонов В-лимфоцитов. По крайней мере, ослабление Т-клеточного иммунитета (иммунодепрессанты, врожденные пороки иммунной системы, СПИД) повышает склонность к развитию ВЭБ-ассоциированных лимфом. В частности, эндемичность лимфомы Беркитта объясняют широким распространением в Центральной Африке и Новой Гвинее малярии, которой сопутствует иммунодепрессия.
С этих же позиций можно представить и патогенез основных симптомов инфекционного мононуклеоза (рис. 16):
- 1) лимфоаденопатию и спленомегалию (результат ВЭБ-индуцированной поликлональной гиперплазии В-лимфоцитов и атакующих их Т-киллеров);
- 2) некротическую ангину (лизис ВЭБ-трансформированных В-лимфоцитов Т-киллерами);
- 3) мононуклеоз — появление в крови множества атипичных (бластоподобных) мононуклеаров (результат активации Т-лимфоцитов ВЭБ-трансформированными В-лимфоцитами; рис. 17);
- 4) образование гетерофильных антител (т.е. антител к «посторонним» антигенам), в частности IgM-агглютининов против бараньих эритроцитов (следствие неспецифической ВЭБ-стимуляции различных клонов антителопродуцирующих клеток).

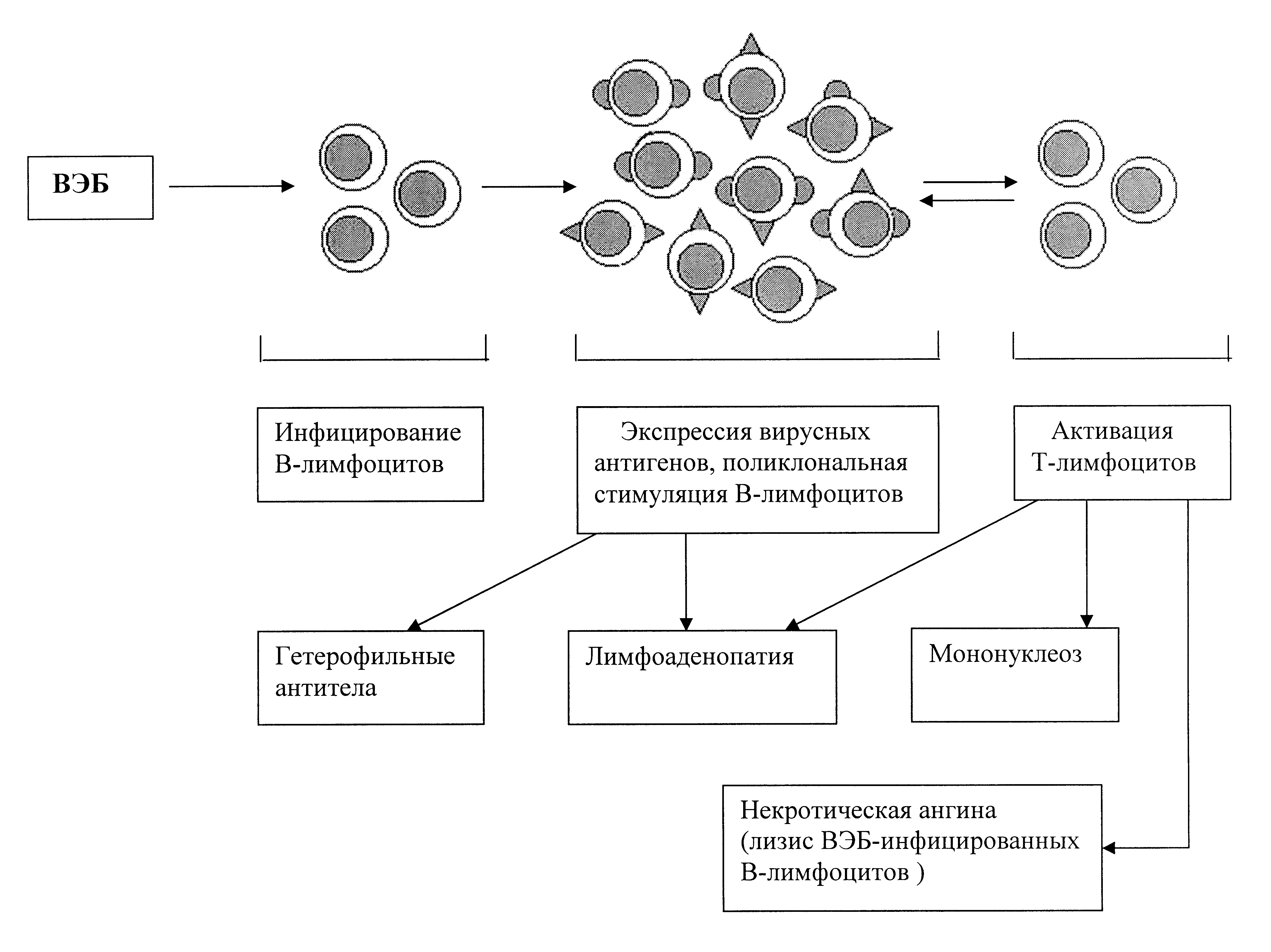

Прогноз — обычно благоприятный, но после выздоровления вирус длительно (несколько месяцев) выделяется из носоглотки. Этиотропная терапия не эффективна, хотя противогерпетические препараты подавляют репликацию ВЭБ in vitro. Это объясняется тем, что патогенез ВЭБ-инфекции связан не с острой репликацией вируса, а с иммунопатологией, индуцированной в ответ на ВЭБ-трансформированные В-лимфоциты.
Клинические проявления реактивации эндогенного ВЭБ не известны. Можно лишь еще раз задуматься над его онкогенными потенциями, прежде всего о связи между ВЭБ-инфекцией и лимфопролиферативными заболеваниями. Проблема неоднозначна, как и вопрос об онкогенности вирусов в целом. В частности, при спорадической лимфоме Беркитта, которая изредка встречается в неэндемичных регионах, ВЭБ обнаруживается лишь в 20% случаев (при почти 100% выделении из эндемичных опухолей). Так же обстоит дело и с В-лимфомами у иммунокомпрометированных больных: не все из них инфицированы вирусом Эпстайна—Барр, как впрочем и другими лимфотропными вирусами.
Вакцинация против ВЭБ отсутствует. При ее разработке акцент должен быть сконцентрирован на изучении вирусных Т-эпитопов для их включения в вакцину (Т-вакцину).
Герпесвирусы человека 6, 7
О широком распространении этих вирусов говорит высокий уровень серопозитивности, который уже в раннем детстве достигает 80—100% (ГВ-6) и 40—80% (ГВ-7). По степени гомологии ДНК они включены в бета-группу цитомегаловирусов, хотя по биологическим свойствам (способность к поражению лимфоцитов) больше похожи на гамма-вирусы.
ГВ-6 впервые изолирован от больных с лимфопролиферативными заболеваниями и даже получил название В-лимфотропного вируса человека. Инфицируя В-лимфоциты, ГВ-6, в отличие от вируса Эпстайна—Барр, не сохраняется в них в латентной форме, а вызывает продуктивную инфекцию, дозревая до вирионов. Клетки, поддерживающие персистенцию вируса, не известны, хотя подозрение падает на CD4 Т-лимфоциты и нейроны ЦНС. Участие ГВ-6 в развитии лимфом поставлено под сомнение. Скорее всего он является причиной широко распространенной детской инфекции — псевдокраснухи (roseola infantum). Она возникает у детей 6 мес — 3 лет при первичном контакте с ГВ-6 и протекает как острое заболевание с сыпью, исчезающей через 3—4 дня после внезапного подъема температуры. Экзантема может отсутствовать, и тогда единственным симптомом является высокая (в течение нескольких дней) температура. Таких случаев, очевидно, немало. Они не диагностируются как герпетическая инфекция и за неимением лучшего воспринимаются как простудные заболевания, хотя местные симптомы ОРЗ отсутствуют. Клинические проявления реактивации ГВ-6 не известны, но есть опасения, что он причастен к развитию множественного склероза и других демиелинизирующих хронических заболеваний ЦНС.
ГВ-7 по своим взаимоотношениям с человеком напоминает ГВ-6. Он тоже ассоциирован с лимфопролиферативными заболеваниями у иммунокомпрометированных больных, но степень его участия в их развитии столь же неопределенна. Наряду с ГВ-6 он считается возбудителем псевдокраснухи. Не исключено, что ГВ-7 персистирует в CD4 Т-лимфоцитах. Клинические проявления латентной инфекции не известны.
Герпесвирус человека 8
Это последний из недавно обнаруженных герпесвирусов человека. Он выделен из СПИД-ассоциированной саркомы Капоши (отсюда его название — Kaposi’s sarcoma-associated herpes-virus, KSHV) и лимфоидных опухолей. Отнесен к гамма-вирусам. Его связь с развитием саркомы Капоши подтвержается присутствием KSHV во всех опухолях данной природы, наличием анти-KSHV-антител и эффективностью антигерпетических препаратов в противорецидивной терапии.
* * * Интерес к герпесвирусам не ослабевает. Этому способствуют острые проявления герпетических инфекций, поиск их фармакологических ингибиторов и возможностей противорецидивной терапии, изучение патогенетического потенциала латентных вирусов, включая их онкогенность и хронические инвалидизирующие процессы. Горьким сюрпризом явилось открытие нового герпесвируса животных. Он явился причиной смертельного геморрагического синдрома у слоненка в Вашингтонском зоопарке и предположительно еще у 8 из 9 слонят, погибших в зоопарках США. Внутренние кровоизлияния возникли у них из-за поражения эндотелия мелких сосудов, главным образом сердца и печени. Это подчеркивает опасения об эндотелиотропности герпесвирусов, в частности, намекает на то, что персистентная герпетическая инфекция каким-то образом связана с атеросклерозом.
Вирусы гриппа
Его ждали по морю на лыжах, а он выкатил из-за печи на кораблях.
- Грипп и ОРВИ.
- История терминов и эпидемий.
- Семейство ортомиксовирусов: А, В, С.
- Субкомпоненты вириона: взаимоотношения с клеткой и организмом.
- Хитрости вирусного генома.
- Патогенез гриппозной инфекции.
- Лидерство гриппа А: причины и следствия.
- Стратегия и тактика изменчивости вируса типа А.
- Эволюция гриппозных вирусов и эпидемиология гриппа.
- Парадоксы иммунитета.
- Профилактика и терапия: желаемое и действительное.
- Грипп В и С.

Внедрение болезнетворных агентов чаще всего происходит через респираторный тракт. Это понятно, если учесть его постоянный контакт с микроорганизмами, взвешенными в ингалируемом воздухе. Некоторые из них высоковирулентны и даже в небольшой концентрации опасны для здоровых людей. Собирательный диагноз ОРЗ (острые респираторные заболевания) подчеркивает полиэтиологичность сходных по клинике поражений респираторного тракта, вызываемых различными возбудителями. Заражению могут подвергаться любые отделы органов дыхания и сопряженные с ними участки тела — придаточные пазухи носа, среднее ухо, конъюнктива. Это ведет к разнообразным клиническим синдромам (рис. 1), причиной которых может быть множество вирусов и бактерий, гораздо реже грибов и простейших.
К инфекциям верхнего отдела респираторного тракта относятся заболевания носоглотки и ротоглотки. В первом случае доминируют назальные симптомы (ринит), часто именуемые малой простудой или просто простудой (англ. cold, или common cold), во втором преобладают признаки фарингита (ангины). Вирусы — единственная причина простуды (40—60% — риновирусы, 15—30% — коронавирусы) и большинства случаев (около 80%) фарингита (риновирусы, коронавирусы, парамиксовирусы, аденовирусы, энтеровирусы). Острые инфекции гортани и трахеи (ларингит, ларинготрахеит) почти всегда имеют вирусную природу (чаще вирусы парагриппа). У детей младшего возраста они нередко проявляются в виде крупа (асфиксии), у взрослых ограничиваются осиплостью голоса и лающим кашлем.
Инфекции нижних отделов респираторного тракта включают поражения бронхов (бронхит), бронхиол (бронхиолит) и легочной паренхимы (пневмония). Бронхиолит (он наблюдается у младенцев) — почти исключительная монополия вирусов, прежде всего респираторно-синцитиального вируса. Этиологический профиль пневмоний неоднороден: вирусы (чаще парамиксовирусы) доминируют в раннем детстве. По мере взросления на первый план выходят бактерии — пневмококки, микоплазмы (M. pneumoniae), хламидии (Сh. pneumoniae), палочка инфлюенцы (Haemophilus influenzae). Речь идет о внебольничных пневмониях. Этиология госпитальных пневмоний выглядит иначе: здесь доминируют возбудители бактериальных оппортунистических инфекций (энтеробактерии, золотистый стафилококк, синегнойная палочка и др.).
ОРЗ-инфекции параназальных синусов и среднего уха обычно имеют бактериальное происхождение (чаще пневмококк, палочка инфлюенцы, моракселлы). Впрочем, и здесь вирусы нередко запускают процесс, который подхватывается бактериями и ведет к вторичным осложнениям. Так возникают многие случаи пиогенного (гнойного) синусита и среднего отита, когда вирусиндуцированный отек слизистой оболочки носа и носоглотки закрывает выходные отверстия придаточных пазух носа и евстахиевой трубы, препятствуя оттоку слизи. Предрасполагающим фактором служит и повреждение реснитчатого эпителия, что ведет к ослаблению естественного очищения слизистых оболочек от бактерий. Особенно страдают дети, у которых ОРЗ наблюдаются чаще и протекают тяжелее, чем у взрослых. Вероятна также прямая вирусная атака, тем более что синусы и среднее ухо выстланы реснитчатым эпителием, чувствительным к респираторным вирусам. По этой же причине поражается и конъюнктива глаза (аденовирусы).
ОРЗ вирусной этиологии выделяют в группу острых респираторных вирусных инфекций — ОРВИ. Это весьма расплывчатая категория, объединяющая инфекции, связанные с так называемыми респираторными вирусами. Их число, особенно с учетом внутривидовых сероваров, впечатляет (одних только риновирусов более сотни). В общей структуре ОРВИ лидирующие позиции принадлежат риновирусам, коронавирусам, вирусам гриппа, парагриппа, респираторно-синцитиальному вирусу. Далее следуют аденовирусы и реовирусы, хотя и они не исчерпывают этиологии ОРВИ.
Заражение ОРВИ-вирусами почти всегда вызывает иммунный ответ. Это говорит о высокой восприимчивости к ним человека и неизбежности конфликта с хозяином. Но иммунитет, индуцируемый таким противоборством, относителен. Инфекции возникают на фоне ощутимых количеств сывороточных и даже мукозальных антител, хотя и протекают менее интенсивно. Возможно, для поддержания резистентности требуется периодическая рестимуляция в виде субклинических и малосимптомных инфекций, и без такой подпитки иммунитет быстро слабеет, чему способствует и антигенное своеобразие возбудителей. Многие новорожденные получают материнские антитела. Но в результате естественного катаболизма их содержание в сыворотке быстро падает (ежемесячно на 50%) и к 2—4 мес снижается ниже протективного уровня. Впрочем, это необязательное условие: клинически значимые инфекции возможны и до истечения «срока годности» пассивного (материнского) иммунитета. К общим признакам ОРВИ относятся преобладание и более тяжелое течение заболеваний у детей младших возрастных групп, повышенная заболеваемость в зимние месяцы и широкая циркуляция возбудителей среди здоровых людей.
Гриппозным вирусам принадлежит особое место в происхождении ОРВИ. Оно определяется уникальной эпидемиологией гриппа, а именно склонностью к крупным вспышкам (эпидемиям и даже пандемиям). Если не бояться парадоксов, грипп следует считать скорее эпидемиологическим, чем клиническим понятием. Именно благодаря своей эпидемической агрессивности он получил статус самостоятельной нозологической единицы. По усредненным симптомам грипп трудно отличить от других ОРВИ, и только массовый характер заболеваемости гарантирует клинический диагноз. Вне эпидемической ситуации определенность вносят лишь специфические методы лабораторного анализа.
Слово «грипп» заимствовано из французского языка и в понятии «эпидемический (заразный) насморк» используется в России с начала ХIХ в. Значительно раньше болезнь стали называть «инфлюэнцей». Это было сделано средневековыми итальянцами, которые полагали, что гриппозные вспышки возникают не без злого влияния небесных тел (в астрологии «influentia» означает эманацию энергии звезд; лат. influere— вливать). Термин «инфлюэнца» принят во многих странах, но у нас утвердился «грипп»— меткое слово, в котором сконцентрирована клинико-эпидемиологическая сущность болезни: острейшее начало, не менее острое развитие и быстрое распространение среди людей (фр. grippе — хватать, схватывать).
Вирусы гриппа беспокоили человечество с античных времен. О заболевании, похожем на грипп, писал Гиппократ, а достоверный счет гриппозным эпидемиям в Европе ведется с 1173 г. Первая из наиболее полно описанных эпидемий относится к 1510 г. За ней последовали вспышки 1557, 1675, 1729—1730, 1742—1743 и 1780 гг. В Х1Х в. также было несколько гриппозных эпидемий, из которых самые значительные относятся к 1831, 1836, 1857—1858, 1874—1875 и, особенно, 1889—1892 гг. Резюмирующий комментарий по этому поводу и сегодня звучит актуально: «В последнюю эпидемию в Петербурге тщательное распространение инфлюэнцы было заметно всякому и без статистических данных. По счастью, эта повальная болезнь не давала у нас большой смертности; но если перевести на деньги количество отнимаемых ею рабочих дней, то оказалось бы, что она обходится человечеству гораздо дороже всякой другой эпидемии, даже холеры. Впрочем, инфлюэнца и сама похищает немало человеческих жизней, а присоединяясь к другим болезням, значительно повышает общую смертность во время своего господства».
В 1918 г. случилась необычная по масштабам и последствиям пандемия (глобальная эпидемия) гриппа. Как и большинство крупных вспышек, она возникла в Юго-Восточной Азии (Китае), но по случайному стечению обстоятельств зафиксирована в истории как «испанский грипп», или просто «испанка». Дело в том, что к этому моменту еще продолжалась первая мировая война и публикации об эпидемиях в воюющих странах были запрещены. Поэтому первое официальное сообщение о тяжелом гриппе вышло из нейтральной Испании. Эпидемий подобного размаха человечество не помнит. В течение 2—3 лет от испанки погибло более 20 млн. человек, и по абсолютному числу смертельных исходов даже за чумой не числится таких рекордов. Вскоре вирус смягчился, и последующие эпидемии гриппа, хотя и перерастали неоднократно в пандемии, не приносили столько жертв. Тем не менее, по показателям летальности грипп продолжает лидировать среди инфекционных заболеваний.
Поиски возбудителя гриппа начались во время эпидемии 1889—1892 гг. Они велись традиционными для бактериологии методами и были заранее обречены на неудачу. О вирусах ничего не знали и искать их целенаправленно никто не мог. И тем не менее «возбудитель» был найден. Его провозгласил Р. Пфейфер, выделивший от больных гриппом палочку инфлюэнцы (см. «Палочка инфлюэнцы»). Тотчас возникли противоречия, но за неимением лучшего до пандемии 1918 г. позицию Пфейфера разделяло большинство микробиологов. «Испанка» окончательно вскрыла ее несостоятельность, и вопрос об этиологии гриппа вновь стал открытым. Первые попытки найти вирус не увенчались успехом, и единственное, что было сделано — собраны и законсервированы сыворотки от переболевших. Они сыграли большую роль в ретроспективной расшифровке этиологии гриппа и развитии представлений об эволюции возбудителя.



Рис. 2. Классическая экспериментальная модель гриппозной инфекции (симптомы ОРЗ): а — здоровый хорек;
б — хорек, зараженный интраназально вирусом гриппа А
Современная история гриппа началась в 1933 г. Зимой этого года был выделен главный из его возбудителей — вирус типа (вида) А. Это удалось сделать В. Смиту и его коллегам при заражении африканских хорьков ультрафильтратом смыва из зева больного гриппом (им был В. Смит — один из авторов открытия). Заболевание, которое последовало у зараженных животных, очень напоминало грипп человека (рис. 2). Круг сомкнулся, когда при помощи материала, полученного от больных животных, грипп воспроизвели на добровольцах. Вирус оказался неприхотливым, и его быстро научились размножать — сначала в куриных эмбрионах, а позже в культурах клеток. В 1940 г. был изолирован вирус В, а в 1947 г. — вирус С.
Общая характеристика вирусов гриппа
Виды (типы)
Вирусы гриппа относятся к семейству ортомиксовирусов, его единственному роду Influenza. Понятие «миксовирусы» было первоначально введено для группы оболочечных РНК-вирусов, которые обладают способностью адсорбироваться на муко(глико)протеиновых рецепторах эритроцитов и вызывать их агглютинацию (гемагглютинация). Название отражает сродство вирусов к муцинам секретов слизистых оболочек (греч. myxa — cлизь). Открытие базисных различий между вирусами гриппа и другими миксовирусами привело к выделению двух семейств — орто- и парамиксовирусов. Парамиксовирусы человека включают вирусы парагриппа, паротита, кори, респираторно-синцитиальный вирус и метапарамиксовирусы (см. «Парамиксовирусы»).
Вирусы гриппа подразделяются на три вида, которые принято называть типами — А, В и С. Иногда их относят к разным родовым таксонам, называя вирусы типов А и В — Influenzavirus А/В, а вирус типа С — Influenzavirus C. Они отличаются по ряду структурных и экологических признаков (см. таблицу), но на практике их дифференцируют по антигенным особенностям нуклеопротеина, который (как, впрочем, и остальные антигены вириона) не дает межтиповых перекрестных реакций, но идентичен у всех штаммов одного типа.
Сравнительная характеристика вирусов гриппа типов А, В и С
| Признак | Вирус А | Вирус В | Вирус С |
| Первое выделение вируса от человека | 1933 г. | 1940 г. | 1947 г. |
| Серотип нуклеопротеина | А | В | С |
| Субтипы (по гемагглютинину и нейраминидазе) | Да | Нет | Нет |
| Резервуар среди животных | Да | Нет | Нет |
| Способность вызывать: | |||
| пандемии* | +++ | — | — |
| эпидемии* | +++ | ++ | — |
| локальные вспышки* | + | ++ | + |
| Поражаемые возрастные группы | Все | Все | Дети, редко взрослые |
| Тяжесть заболевания | +++ | ++ | + |
| Количество РНК-сегментов | 8 | 8 | 7 |
| Поверхностные антигены: | |||
| гемагглютинин | Да | Да | Да |
| нейраминидаза | Да | Да | Нет |
| Изменчивость поверхностных антигенов: | |||
| интенсивность | +++ | + | +/– |
| шифт | Да | Нет | Нет |
| дрейф | +++ | +/– | |
| Чувствительность к ремантадину/амантантану | Да | Нет | ? |
*Под вспышкой следует понимать групповую заболеваемость в ограниченном организованном коллективе (детское учреждение, учебное заведение, воинская часть, предприятие и т.п.). Под эпидемией — превышение ожидаемого неэпидемического уровня острых респираторных инфекций в городе; эпидемией в стране считаются эпидемии одной этиологии в нескольких городах. Пандемия — ряд последовательных эпидемий одной этиологии в нескольких странах, расположенных в разных частях света.
Безусловный лидер по патогенности — вирус типа А, тип В занимает промежуточное положение, вирус типа С выполняет роль аутсайдера. Но сначала следует сказать о том, что объединяет семейство Orthomyxoviridae.
Вирион
Вирусы гриппа представляют собой частицы плеоморфной (обычно сферической) формы, среднего для вирусов размера (80—120 нм в диаметре). Они содержат РНК, которая упакована в комплексе с белками в виде спирали (кор, или нуклеокапсид) и окружена внешней оболочкой (суперкапсид). На поверхности вирионов имеется множество мелких шипов, построенных из гликопротеинов с гемагглютинирующей или нейраминидазной активностью (рис. 3). У вируса типа С нейраминидаза отсутствует; ее заменяет ацетилэстераза, отрезающая ацетильные группы гликопротеинов.

РНК обладает негативной полярностью (минус-нить). Это означает, что, попав в клетку, она не может напрямую (без транскрипции) выполнять функции мРНК. Но главная особенность генома — фрагментарность. РНК расчленена на 8 (типы А и В) или 7 (С) сегментов, большинство из которых кодируют единственный белок, т.е. являются моноцистронными генами. Один из сегментов кодирует неструктурные протеины (NS1 и NS2), существующие только в инфицированных клетках.
Вирион состоит из 7 белков, которые, как и у всех оболочечных вирусов, делятся на внутренние и поверхностные. К внутренним относятся матриксный белок (М) и белки сердцевины: нуклеопротеин (NP) и ферменты полимеразного комплекса. Поверхностные белки представлены гемагглютинином и нейраминидазой (вирусы А и В).
М-белок расположен на внутренней поверхности суперкапсида, сопрягая его с сердцевиной. Это стабилизирующее начало вириона, и при его отсутствии или неполноценности вирусная частица теряет прочность, быстро распадаясь на субкомпоненты. Нуклеопротеин образует чехол (капсид) вокруг РНК, не утрачивая с ней связи на протяжении всего репликативного цикла. Это не только не мешает экспрессии генов, но даже помогает транскрипции и репликации. Белки РВ1, РВ2 и РА являются ферментами, играя ключевую роль в манипуляциях с вирусными РНК внутри зараженных клеток (см. ниже). РВ1 и РВ2 обладают основными (англ. basic), а РА — кислыми (англ. acidic) свойствами. Это послужило поводом для их названия, В или А.
Все внутренние белки — типоспецифические антигены. Это означает, что их эпитопный профиль радикально отличается у разных типов вируса гриппа, но практически идентичен для всех штаммов каждого из них. Собственно, и само подразделение на типы (виды), как уже говорилось, имеет серологическую основу, базируясь на антигенной дискретности А-, В- и С-нуклеопротеинов. Антитела против внутренних антигенов лишены защитного действия.
Поверхностные белки (гемагглютинин и нейраминидаза) входят в состав суперкапсида. Это лицо вируса, по которому можно отличить близких родственников, судить об их эпидемических претензиях и взаимоотношениях с хозяином. Они заякорены гидрофобными концами в липидном бислое суперкапсида и структурно оформлены в виде шипов, выступающих на поверхности вириона (см. рис. 3). Изнутри гемагглютинин и нейраминидаза контактируют с М-белком, а через него — с сердцевиной.
Гемагглютинин (Н) выполняет рецепторную функцию при заражении клеток. Это объясняется его сродством к сиалированным гликопептидам и гликолипидам, благодаря которому вирионы закрепляются на плазматической мембране. Во взвеси с эритроцитами человека и многих животных вирусные частицы одновременно соединяются с двумя клетками, вызывая гемагглютинацию. Эта простая реакция имеет большое индикаторное значение и широко используется в дифференцировке вирусных штаммов, диагностике гриппа и оценке протективных ресурсов иммунитета.
Но значение гемагглютинина не ограничивается физическими контактами между вирусом и клеткой. Он продолжает действовать и на последующих этапах инфекции, способствуя выходу свободного нуклеокапсида в цитоплазму. Это происходит в кислой среде эндосом (фаголизосом), благодаря обнажению специальных структур гемагглютинина (так называемых сайтов слияния), которые возбуждают объединение вирусной и клеточной мембран. Полифункциональность гриппозного гемагглютинина уникальна: у других вирусов (например, у тех же парамиксовирусов) функции рецепции и слияния поделены между разными белками. Кроме того, гемагглютинин выполняет роль главного протективного антигена: антитела к нему нейтрализуют вирус и подавляют развитие инфекции.
Гемагглютинин синтезируется и включается в состав вириона в виде предшественника, который способен реагировать с рецепторами клеток, но не обеспечивает слияния вирионной оболочки с клеточными мембранами. Для реализации этой важнейшей функции (см. ниже) гемагглютинин должен пройти через этап ограниченного протеолиза. В условиях естественной инфекции его вызывают сериновые протеазы, которые в изобилии содержатся в респираторных секретах. Они расщепляют гемагглютинин на два связанных между собой дисульфидным мостиком фрагмента — Н1 и Н2. После дополнительных конформационных изменений в кислой среде эндосом Н2 внедряется своим гидрофобным концом в липидный бислой клеточной мембраны, инициируя процесс слияния. Без такой поддержки со стороны хозяина вирус биологически инертен. Это одна из причин великолепного размножения вирусов гриппа в курином эмбрионе и культуре клеток птиц, которые очень богаты протеазами, расщепляющими гемагглютинин. В культурах клеток млекопитающих, лишенных протеазной активности, образуются вирионы с нерасщепленным гемагглютинином. Они нормально прикрепляются к клеткам-хозяевам и агглютинируют эритроциты (результат активности Н1), но неинфекционны. Добавление в среду трипсина обеспечивает репликацию вируса и цитопатический эффект.
Нейраминидаза (N) образует отдельные шипы, которых в несколько раз меньше, чем гемагглютинина. Это фермент, отщепляющий N-ацетилнейраминовую (сиаловую) кислоту от гликопептидов и гликолипидов (ганглиозидов), т.е. по сути инактивирующий рецепторы для гемагглютинина. В этом легко убедиться, наблюдая за вирусиндуцированной гемагглютинацией: агрегаты эритроцитов, стабильные при 4оС, быстро распадаются при 37оС, так как нейраминидаза откусывает гемагглютинин вместе с сиаловой кислотой. Нечто подобное происходит и во время инфекционного процесса. Нейраминидаза отделяет вирионы от сиалированных муцинов, покрывающих слизистую оболочку, способствуя продвижению вируса к поверхности эпителиальных клеток. При завершении репликативного цикла она помогает отделению созревших вирионов от эпителиоцитов. И в том и в другом случае нейраминидаза действует как фактор распространения, расширяя зону инфекции. Поэтому антитела к нейраминидазе обладают защитным действием, хотя и более слабым, чем антитела против гемагглютинина. В клеточных культурах они сокращают размер, но не количество вирусных колоний. Это говорит о торможении распространения вируса от клетки к клетке при сохранении числа инфекционных вирусных частиц, т.е. об отсутствии вируснейтрализующего эффекта.
С поверхностными белками (H, N) связана высокая антигенная изменчивость вирусов гриппа. Она определяет рецидивы эпидемической агрессивности вируса типа А, меньше проявляется у вируса типа В и, по-видимому, не имеет практического значения для гриппа С (см. ниже).
Репликация
Вирус проникает в клетку путем эндоцитоза после связывания гемагглютинина с сиалированными клеточными рецепторами. Фагосома (эндосома), в которую заключен вирус, сливается с лизосомами. Кислая среда внутри фаголизосомы меняет конформацию гемагглютинина, обнажая пептиды, вызывающие слияние вирусной и эндосомальной мембран. Благодаря этому нуклеокапсид освобождается от суперкапсида и выходит в цитоплазму. Отсюда вместе с М-белком он быстро транспортируется в ядро, где уже через несколько минут после заражения появляются первые РНК-транскрипты. Число разновидностей мРНК соответствует количеству сегментов геномной РНК, хотя спектр синтезируемых белков несколько больше: к структурным белкам вириона добавляется 3 неструктурных белка, принимающих участие в репликации вируса. Расширение генетической емкости РНК достигается благодаря изящному механизму — повторной трансляции одной и той же мРНК со сдвигом рамки считывания ее генетического кода.
Ортомиксовирусы едва ли не единственные из РНК-вирусов, манипулирующие со своей геномной молекулой в ядре зараженной клетки. Клетки не умеют транскрибировать РНК, поэтому операции такого рода обязан производить сам вирус. Толчок дает полимеразный комплекс вириона, позже начинают работать вновь синтезированные ферменты. Белки РВ1 и РВ2 обеспечивают копирование геномных фрагментов с образованием неполных и полных по длине транскриптов. Укороченные копии выполняют функции мРНК. Для этого они подвергаются доработке, обретая облик, привычный для мРНК клетки (присоединение к 5`-концу шапочки из метилированных нуклеотидов и полиаденилирование 3`-конца). Не располагая собственными ресурсами для прямого решения этой задачи, вирус находит обходной путь. Он подключает свою эндонуклеазу (возможно, белок РА), которая откусывает недостающие фрагменты от мРНК клетки-хозяина и переносит их на вирусные РНК-транскрипты. Столь необычное и тонкое проявление молекулярного паразитизма объясняет природу внутриядерной репликации вирусов гриппа: только здесь они могут найти свежие (еще не связавшиеся с белком) мРНК, чувствительные к вирусной эндонуклеазе.
Полные по длине РНК-транскрипты не модифицируются и поэтому не могут работать как мРНК. Они остаются в ядре и служат матрицей для образования репликативных РНК, идущих на построение новых нуклеокапсидов. В этом принимает участие РНК-полимераза, образующаяся при помощи первых мРНК.
Готовые мРНК переправляются в цитоплазму, где транслируются на рибосомах в вирусные белки. Часть синтезированных белков вновь переходят в ядро и после комплексации с репликативными РНК образуют нуклеокапсиды. Гемагглютинин и нейраминидаза включаются в плазматическую мембрану, готовя фрагменты будущего суперкапсида. С внутренней стороны липидного бислоя к ним присоединяется М-белок, который ориентирует закрепление нуклеокапсидов в зонах почкования новых вирионов. При отделении от клеток вирус получает суперкапсид с его важнейшими атрибутами — гемагглютинином и нейраминидазой (рис. 4, 5).
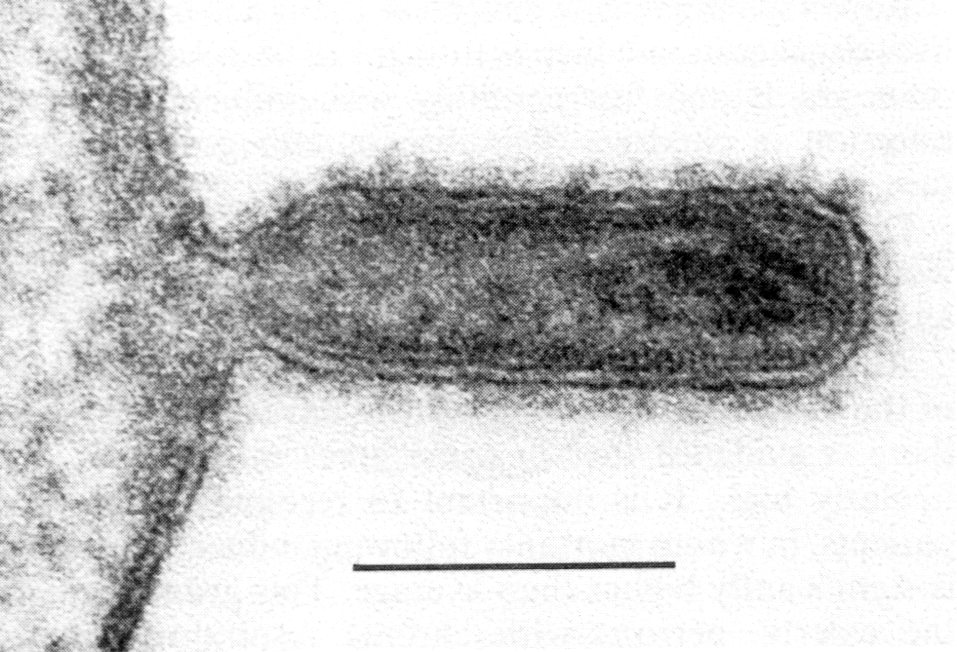


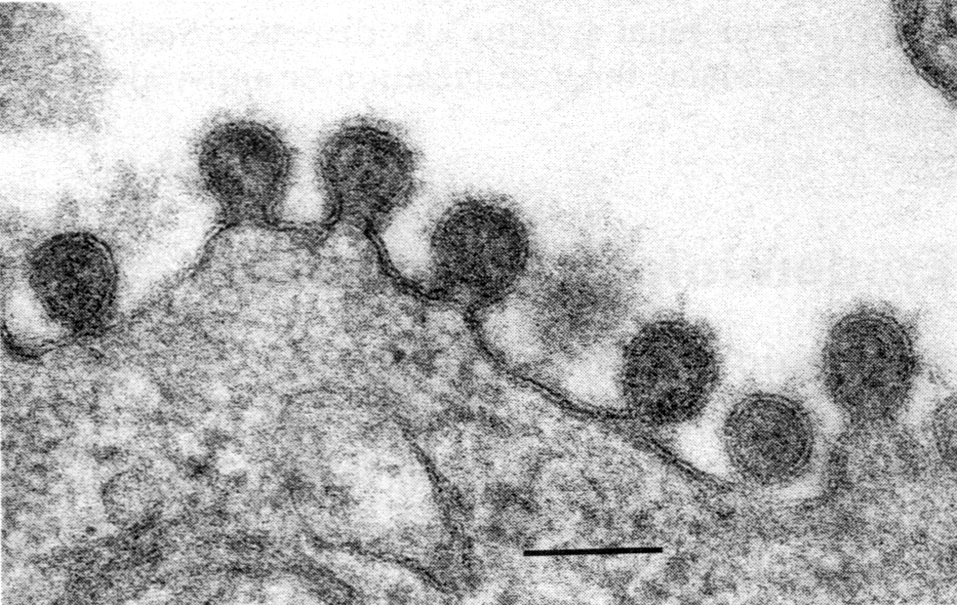
соответствует 100 нм

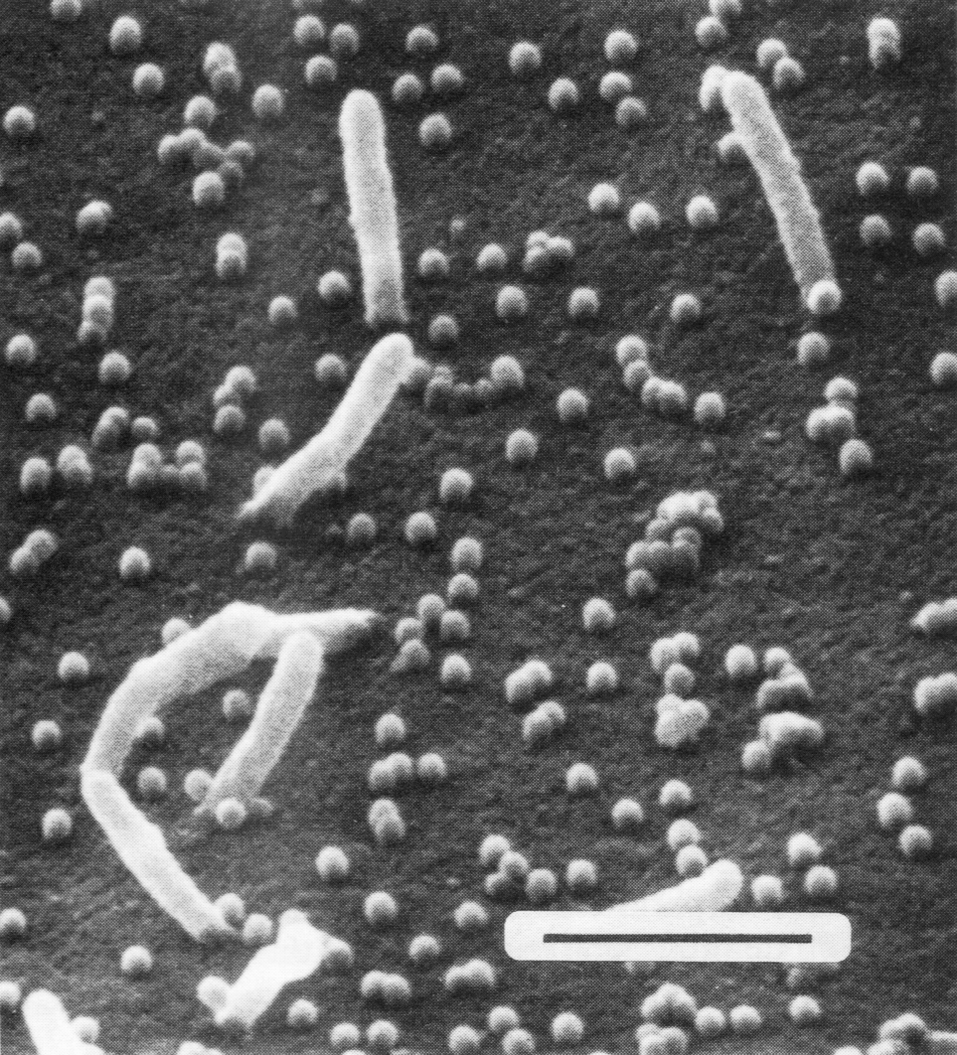
Как уже говорилось, гемагглютинин синтезируется в виде предшественника, который активируется клеточными протеазами (здесь корень формальной логики, породившей иллюзию о возможности лечения вирусных инфекций банальными ингибиторами протеаз). Сбрасывание созревших вирионов и их распространение на соседние клетки усиливается нейраминидазой. Репликация в чувствительных клетках происходит очень быстро, и уже через 6—8 ч в среде появляется вирусное потомство.
Экология
Кроме человека в естественных условиях вирусы гриппа инфицируют млекопитающих и птиц. Еще в 1931 г., т.е. за два года до открытия человеческого вируса, Р. Шоуп выделил пневмотропный вирус при массовых заболеваниях свиней. Тогда никто не предполагал, что это отпрыск возбудителя страшной «испанки», адаптировавшийся к новому хозяину во время пандемии 1918—1919 гг. Более того, оказалось, что вирус чумы домашних птиц (она протекает в виде септицемии, т.е. клинически совсем не похожа на грипп человека), обнаруженный в 1901 г., тоже близкий родственник вирусов гриппа человека. Эти открытия были оценены по достоинству с опозданием, когда появилась возможность серо- и генотипирования вирусных штаммов. Вирусы гриппа изолированы от лошадей, китов и тюленей. Обширным резервуаром служат птицы, выделяющие вирус с испражнениями.
Все изоляты от животных относятся к типу А. Они не патогенны для людей, но играют важную роль в эволюции эпидемических штаммов. Справедливо и обратное. Вирусы человека не вызывают эпизоотий, хотя пример с «испанским гриппом» свиней не исключает такой возможности. В эксперименте повышение вирулентности гриппозных штаммов наблюдается уже после нескольких пассажей на животных. Адаптация носит избирательный характер, распространяясь на узкий круг родственных хозяев.
Впрочем, в последние годы зарегистрированы случаи заражения людей птичьими штаммами вируса А. Реальная тревога возникла в 1997 г. в Гонконге, когда 18 человек напрямую заразились от домашних птиц вирусом H5N1; 6 человек погибло. В 2003 г. высоковирулентные штаммы птиц H5N1 и H7N7 спровоцировали фатальные случаи гриппа среди жителей Южной Азии. К счастью, они не получили распространения, но тем не менее очертили круг задач, решение которых необходимо для профилактики новых гриппозных пандемий.
Вирусы типов В и С являются сугубо человеческими и в естественных условиях не инфицируют других хозяев.
Патогенез
Вирусы гриппа проникают в респираторный тракт с каплями аэрозоля и частицами пыли. Чем мельче их величина, тем глубже проникает вирус; самые мелкие частицы достигают бронхиол и альвеол. Главным местом размножения вируса служат реснитчатые и бокаловидные клетки слизистой оболочки верхних дыхательных путей, но инфекция может охватить весь респираторный тракт — от носовых ходов до терминальных бронхиол и альвеол. Благодаря короткому репликативному циклу из каждой вирусной частицы к концу первых суток возникает огромное потомство — 1027. Это объясняет короткий инкубационный период (1—3 сут) и острейшее начало гриппа.
Повреждение зараженных эпителиоцитов индуцирует воспалительную реакцию, в которой преобладают макрофаги и лимфоциты. Подвергаясь активации, они секретируют множество флогогенных начал (прежде всего цитокинов), которые вызывают общую интоксикацию и поддерживают местный воспалительный процесс. В типичных случаях после внезапного недомогания, озноба с быстрым повышением температуры до 38—40оС, головной и часто мышечных болей развиваются симптомы поражения верхних (ринит, фарингит) и глубжележащих (ларингит, трахеобронхит) дыхательных путей. Общая интоксикация характерна для гриппа, но часто доминируют местные проявления. Наиболее тяжело протекает грипп А (хотя история эпидемий убеждает в вариабельности этого признака); вирусы типа В и, особенно, С менее агрессивны. В целом, опираясь только на клинику, отличить грипп от других ОРЗ практически невозможно. Без лабораторного подтверждения это удается лишь в сочетании с эпидемиологическим диагнозом, на фоне вспышки гриппа среди населения.
Вирус редко проникает в кровь. Это замечено лишь в тяжелых случаях, но вряд ли имеет патогенетическое значение, хотя бы из-за отсутствия сывороточных и тканевых протеаз, способных активировать вирусный гемагглютинин. Казуистикой вирусемии можно объяснить и отсутствие тератогенных последствий гриппозной инфекции, возникшей во время беременности. Достоверные данные на этот счет отсутствуют.
Из осложнений наиболее опасна вторичная бактериальная бронхопневмония. Она развивается в зонах выраженного повреждения эпителия, чаще у пожилых людей, особенно при хронических заболеваниях дыхательной и сердечно-сосудистой систем. Это главная причина летальных (порой катастрофически быстрых) исходов. Возбудителями чаще служат инкапсулированные бактерии (пневмококк, палочка инфлюэнцы), но опасны и другие бактерии, прежде всего золотистый стафилококк. Симптомы (лихорадка, кашель с гнойной мокротой, хрипы и пр.) обычно возникают в виде второй волны, в период реконвалесценции от гриппа. Гораздо реже встречается первичная пневмония, вызываемая самим вирусом гриппа. Иногда она обретает геморрагический характер (мокрота с кровью) на фоне массивного повреждения эндотелия и в таких случаях быстро ведет к смерти. Это было характерно для «испанского гриппа», когда многие больные буквально задыхались в собственной сыворотке или крови в течение 48 ч.
Освобождение от вируса начинается тотчас после повреждения слизистой оболочки. Продукты распада эпителиоцитов и медиаторы воспаления стимулируют продукцию слизи, а уцелевшие клетки мерцательного эпителия и кашлевые толчки проталкивают ее вверх вместе с основной массой вируса. Этот простой, но надежный механизм в сочетании с ринореей предупреждает распространение инфекции по респираторному тракту, способствуя выздоровлению. Во всяком случае, клиническое улучшение наступает раньше, чем в крови или секретах обнаруживаются противовирусные антитела, хотя полное освобождение от вируса совпадает с появлением секреторных антител. В неосложненных случаях больной поправляется через 7—10 дней.
Грипп А
С вирусом типа (вида) А связаны главные проблемы гриппозной инфекции. Это эталон, фокусирующий все, что на сегодня известно об ортомиксовирусах, эпидемиологии, иммунологии и клинике гриппа.
Классификация
Выраженная изменчивость вируса гриппа А потребовала унифицировать внутритиповую классификацию штаммов на основе антигенных особенностей гемагглютинина и нейраминидазы. С 1980 г. все известные разновидности гемагглютинина сгруппированы в 13 (Н1—Н13), а нейраминидазы — в 9 субтипов (N1—N9).
Современная классификация состоит из обозначения штаммов и описания субтипов гемагглютинина и нейраминидазы. Обозначение штаммов включает пять позиций: 1) тип (А, В и С); 2) естественный хозяин, если это не человек; 3) географическое происхождение; 4) порядковый номер; 5) год выделения. Антигенная характеристика гемагглютинина и нейраминидазы следует в круглых скобках за обозначением штамма. Примеры: А/Гонконг/1/68(H3N2); А/Токио/3/67(Н2N2); А/СССР/90/77(H1N1); A/утка/СССР/695/76(H3N2).
В формуле вирусов В и С описание H- и N-антигенов отсутствует.
Антигенная изменчивость
Вирусы гриппа А обладают удивительно нестабильным геномом, вариабельность которого на 2—3 порядка выше, чем у большинства других РНК-вирусов. Изменчивость затрагивает все компоненты вируса, но ее главные проявления связаны с поверхностными структурами — гемагглютинином и нейраминидазой. Именно их эпитопные перестройки определяют антигенную формулу штаммов и лежат в основе эпидемиологии гриппозной инфекции. Причина высокой вариабельности поверхностных антигенов при относительном постоянстве внутренних компонентов (нуклеопротеина, M-белка, полимеразного комплекса) связана с отбором иммунорезистентных вирусных клонов. Поскольку внутренние структуры экранированы от антител, селекции и избирательной экспансии подвергаются прежде всего антигенные варианты суперкапсида. Эпитопные изменения внутренних белков не создают преимуществ во взаимоотношениях с хозяином и обычно не закрепляются в вирусной популяции. Чтобы это произошло, структурные перестройки белков должны позитивно влиять на функции, от которых зависит лидерство вируса в инфекционном процессе.
Примером такого рода выгодного для вируса сочетания иммунорезистентности по внешним антигенам и высокой вирулентности (она зависит от взаимодействия всего комплекса вирусных белков) являются эпидемические штаммы, которые периодически получают распространение среди людей (см. ниже).
Гемагглютинин и нейраминидаза меняются независимо друг от друга, благодаря двум генетическим механизмам — шифту и дрейфу. Шифт (англ. shift — cдвиг) ведет к полной смене антигенного профиля молекул H/N и появлению новых субтипов вируса. Дрейф (англ. drift — медленное течение) означает частичное обновление H/N-эпитопов при сохранении антигенного родства с поверхностными антигенами прототипного (родительского) штамма. Благодаря этому в изначально гомогенной вирусной популяции возникают клоны, которые, подвергаясь селекции, дают начало штаммам с повышенной эпидемической проходимостью. Иными словами, если шифт порождает новые субтипы гемагглютинина и нейраминидазы, то дрейф определяет штаммовые особенности внутри каждого субтипа. Шифт — стратегия, дрейф — тактика антигенной изменчивости.
Новые шифт-варианты (субтипы) возникают редко. Для вирусов человека за последние сто лет зарегистрировано только три шифта по гемагглютинину (Н1, Н2, Н3) и два — по нейраминидазе (N1, N2). Всего известно 13 шифтовых вариантов гемагглютинина и 9 — нейраминидазы. Они существуют в различных комбинациях, большинство из которых выделяется от животных. Наибольшая мозаика антигенных вариантов выявлена у диких и домашних птиц. Их вирусы нередко сочетают оригинальные гемагглютинины с нейраминидазами вирусов гриппа млекопитающих или людей, позволяя размышлять о возможности происхождения новых субтипов на основе генетических пересортировок между штаммами разной природы. Что касается вирусов человека, то начиная с 1918 г. они были представлены лишь тремя антигенными формулами: H1N1, H2N2, H3N2.
В отличие от шифтов, возникающих неожиданно и скачкообразно, дрейф совершается непрерывно, как многоступенчатый процесс. Поэтому дрейф-варианты сохраняют антигенную преемственность, которая снижается по мере накопления структурных перестроек. В конце концов антигенное родство может стать минимальным, приближаясь к шифту. Однако благодаря эстафетности мелких изменений (достаточно замены единственной аминокислоты) все дрейф-варианты представляют единую семью, члены которой более или менее похожи по гемагглютинину и нейраминидазе.
Дрейф определяется точечными мутациями в генах гемагглютинина и нейраминидазы. Это соответствует повышенной мутабельности РНК-вирусов, которая связана со сравнительно низкой надежностью РНК-реплицирующих ферментов и неспособностью клеток исправлять их ошибки. Вопрос лишь в том, почему вероятность дрейфа у вируса гриппа гораздо выше, чем у других РНК-вирусов? Вполне убедительного объяснения не существует. Одной из причин может быть экологическая привязка гриппозных вирусов к респираторному тракту. IgA-антитела, с которыми вирус сталкивается в верхних дыхательных путях, образуют непрочные комплексы с антигенами и не очень надежно нейтрализуют вирусы. Это позволяет вирусу активно размножаться, расширяя свою популяцию, а следовательно, и вероятность накопления мутаций. Некоторые из мутантов могут быть еще резистентнее к секреторным антителам, создавая прецедент для селекции. Это согласуется с наблюдениями о слабой антигенной изменчивости тех РНК-вирусов, которые обязаны пройти через этап вирусемии (например, вирусы кори, паротита). В отличие от вирусов, не покидающих слизистые оболочки, они эффективно нейтрализуются циркулирующими IgG-антителами, не успевая накопить потомство, обогащенное мутациями. Впрочем, это не объясняет высокой изменчивости вируса гриппа А относительно других респираторных РНК-вирусов (парамиксовирусов, вирусов гриппа В и С и пр.).
О механизме шифтовой изменчивости продолжают спорить, но большинство склоняется к тому, что главной причиной является перетасовка (рекомбинация) генов при одновременном заражении клеток несколькими штаммами вируса. Наиболее удачливые рекомбинанты избегают нейтрализующего действия антител, получая шанс для выживания. Вероятность генетических перетасовок возрастает благодаря фрагментарности вирусного генома, которая обеспечивает обмен целыми генами, кодирующими дискретные вирусные белки.
Источником новых шифт-вариантов могут быть животные, прежде всего птицы. Они заражаются вирусами человека, обеспечивая их взаимодействие с собственными штаммами. Считают, например, что вирус Гонконг (H3N2), который появился в 1968 г., открыв современный пандемический цикл, представляет собой гибрид, сохранивший 7 генов своего предшественника (азиатского вируса H2N2) и получивший новый ген для гемагглютинина (Н3) от вируса животных. В лабораторных условиях вирусы человека и животных охотно вступают в генетические перетасовки, давая гибридное потомство. В отличие от вируса типа А, вирусы типов В и С лишены шифтовой изменчивости. Это совпадает с отсутствием у них внечеловеческого резервуара в природе.
Эпидемиология
Эпидемиологию гриппа А определяют три главных фактора:
- высокая чувствительность человека к возбудителю (достаточно несколько вирионов, чтобы вызвать заболевание у неиммунных людей);
- широкие контакты с вирусом благодаря воздушно-капельному способу заражения;
- антигенная изменчивость вируса, которая лишает надежности и даже сводит на нет анамнестический иммунитет, приобретенный в результате прошлых контактов с вирусом.
Хотя в природе существует обширный резервуар вирусов гриппа А среди животных, они не представляют (пока!) эпидемической угрозы для человека. Штаммы, болезнетворные для людей, столь же избирательно адаптированы к человеку, как вирусы гриппа млекопитающих или птиц к своим хозяевам. Единственным источником инфекции служит человек — больные гриппом, реже здоровые вирусоносители. Наиболее опасны больные стертыми формами заболевания.
Вирус гриппа А знаменит благодаря своей способности вызывать эпидемии, которые могут перерастать в пандемии, охватывая значительную часть населения Земного шара. Впрочем, если учесть, что современный грипп — непрерывный процесс, и в глобальном масштабе постоянно регистрируются более или менее обширные вспышки заболевания, следует говорить скорее о пандемических циклах, подразумевая цепь последовательных эпидемий в различных странах. Начиная с 1918 г. отмечено не более 4 пандемических циклов, каждый из которых был связан с новым шифт-вариантом (субтипом) (табл. 2).
Таблица 2
Пандемические циклы гриппа А
| Антигенная формула вируса | Продолжительность цикла |
| H1N1 | 1918–1957 |
| H2N2 | 1958–1967 |
| H3N2 | 1968– |
| H1N1 | 1977– |
Периодические (через 1—3 года) подъемы заболеваемости внутри пандемических циклов объясняются дрейфом суперкапсидных антигенов (прежде всего гемагглютинина), который позволяет вирусу избегать анамнестических антител, накопившихся в популяции после контактов со штаммами-предшественниками. Это означает, что при прочих равных условиях именно коллективный иммунитет селекционирует поверхностные структуры, обновляемые в результате антигенного дрейфа, одновременно отторгая консервативные (т.е. сохранившие прежние эпитопы) вирионы.
Вспышка гаснет по мере расширения иммунной прослойки (в большом городе на это уходит около месяца), но при нынешней скорости коммуникаций, интенсивности деловых поездок и туризма обновленный вирус успевает перекочевать на соседние территории и многократно разыграть сходный сюжет. Есть даже мнение, что в глобальном масштабе у гриппа А нет межэпидемического периода, и «светлые промежутки» носят региональный характер. Но коллективный иммунитет в конце концов берет верх над эволюцией вируса. После серии эпидемий, связанных с дрейф-вариантами, он обретает силу, достаточную для вытеснения субтипа, возбудившего пандемический цикл. История учит, что для этого требуется 10—40 лет. Место потерявшего актуальность вируса занимает новый субтип — продукт радикальных (шифт) вариаций гемагглютинина и нейраминидазы. Он запускает следующий пандемический цикл, который обычно стартует из густо населенных регионов Юго-Восточной Азии и быстро втягивает в глобальный эпидемический процесс другие территории Земного шара. После первой, наиболее обширной и тяжелой волны гриппа следует длительное противостояние дрейф-мутантам, которое после серии эпидемических потрясений завершается элиминацией пандемического подтипа из человеческой популяции и освобождением места для очередного (не похожего на предыдущие) шифт-варианта.
Эта классическая схема была подпорчена в мае 1977 г., когда в северных регионах Китая появился субтип Н1N1, т.е. тот же вариант, который стал причиной трагической пандемии «испанки» 1918—1919 гг. и лишь в 1957 г. был замещен вирусом азиатского гриппа (H2N2). Возвращение испанского субтипа явилось неожиданностью, так как возможность рециркуляции исчезнувших гриппозных сероваров большинством авторов отрицалась. Кроме того, в это время господствовал гонконгский вирус с антигенной формулой H3N2 (он вытеснил азиатский вирус в 1968 г.), и по логике циркуляция других сероваров исключалась. Стали ждать тяжелых последствий, памятуя о жертвах «испанки». Инфекция распространилась по всему миру, но, к счастью, протекала легко, причем в основном болели лица моложе 20 лет, т.е. те, кто родился после 1957 г., когда вирус Н1N1 перестал выделяться от людей. Не известно, где и как он прятался после первого пандемического цикла. Эта неопределенность вылилась даже в подозрение о том, что по небрежности возбудитель выскользнул из какой-то лаборатории, запутав и без того противоречивый сюжет о природной эволюции вирусов гриппа. Но так или иначе с тех пор (т.е. с 1977 г. по настоящее время) в пандемическом цикле задействованы два субтипа — H3N2 и H1N1. Более того, были выделены штаммы-рекомбинанты, содержащие гены обоих вирусов, что вызвало новую тревогу, но опасения об их повышенной вирулентности не оправдались.
Гораздо меньшая агрессивность субтипа H1N1 образца 1977 г. по сравнению с аналогичным серотипом 1918 г. навела на размышления о том, что вирулентность вируса не исчерпывается антигенными свойствами поверхностных структур, а уклонение от антител — необходимый, но недостаточный механизм болезнетворности. Эпидемичность (а тем более пандемичность) штаммов зависит и от других признаков, которые делают вирус высококонтагиозным и цитопатичным. Об этом же говорит и неодинаковая болезнетворность вирусных изолятов в масштабе одной эпидемии, несмотря на идентичность антигенной формулы. Можно вспомнить и о безвредности для людей гриппозных вирусов животных, хотя по функциональным свойствам гемагглютинина и нейраминидазы они мало чем отличаются от вирусов человека. Факторы, делающие штамм предельно вирулентным (и в этом смысле дополняющие поверхностные компоненты), не известны, но они тоже связаны с изменчивостью вируса, участвуя в его эволюции через взаимодействие с популяцией чувствительных хозяев и их клеток. В целом эпидемичность гриппозных вирусов — полигенный признак, который формируется в результате сочетанных мутаций и межштаммовой перетасовки генов, кодирующих разные вирусные белки, среди которых наиболее очевидную роль играют вариации по поверхностным антигенам.
Более понятен механизм ослабления вирулентности. Вирус достаточно охотно идет на это, и в его популяциях всегда присутствуют варианты со сниженной болезнетворностью или лишенные ее. К ним, например, относятся холодолюбивые мутанты, адаптированные к пониженной (30—37оС) температуре, и дефектные интерферирующие частицы, которые мешают репродукции стандартного вируса, сокращая инфекционное потомство. Причиной дефектности является утрата или повреждение генов, кодирующих белки полимеразного комплекса. Не исключено, что ослабленные клоны способствуют персистенции вируса в биологических системах, создавая прецедент для его реактивации и рециркуляции. В это продолжают верить некоторые вирусологи, хотя как закономерное явление длительное бессимптомное носительство вируса гриппа А известно только у птиц.
Иммунитет
Каждый из нас обладает определенным запасом резистентности против вирусов гриппа. Она неспецифична, реализуется на уровне слизистых оболочек респираторного тракта (механические факторы и мукоцилиарный транспорт, противовирусные ингибиторы секретов, интерфероны) и обеспечивает устойчивость к заражению или по крайней мере бессимптомное течение инфекции у 20—50% людей, впервые столкнувшихся с вирусом. Те же механизмы действуют и при клинически манифестной инфекции, выполняя большой объем черновой работы по уничтожению вируса.
Главная роль в превентивном иммунитете и завершении вирусной стерилизации дыхательного тракта принадлежит специфическому иммунитету, прежде всего антителам респираторных секретов. Гриппозная инфекция сопровождается образованием антител ко всем структурным и неструктурным белкам вируса, однако протективными свойствами обладают только антитела к гемагглютинину и нейраминидазе, причем решающее значение имеют гемагглютининовые антитела. Именно они служат критерием устойчивости человека к гриппозной инфекции, отвечая за эффективность постинфекционного и поствакцинального иммунитета. Наиболее активны антитела к штаммоспецифическим эпитопам гемагглютинина; групповые эпитопы, общие для всех штаммов данного субтипа, если и индуцируют синтез антител, то только слабых, неэффективных.
Протективность проще всего связать с подавлением рецепции вирионов на клетках, хотя с учетом полифункциональности гемагглютинина механизм вируснейтрализующего эффекта направленных против него антител может быть сложнее. В частности, некоторые из вирионов, покрытых антителами, проникают в клетки, но лишаются способности к транскрипции своей РНК.
Антитела к нейраминидазе менее надежны. Тем не менее, и они вносят существенный вклад в защиту от вируса, препятствуя диссеминации инфекции по респираторному тракту. Это ограничивает поражение глубоких дыхательных путей, снижая тяжесть заболевания и выделение вируса во внешнюю среду.
IgA-антитела синтезируются местно и обеспечивают защиту верхних дыхательных путей. Иммунитет бронхов и легких связан в основном с IgG-антителами, поступающими из крови. Это объясняет корреляцию между титром гемагглютининовых антител сыворотки и резистентностью к инфекции — титр 1:40 (в реакции торможения гемагглютинации) считается достаточным для защиты от эпидемического (т.е. актуального на данный момент) штамма.
Гриппозной инфекции сопутствует появление противовирусных цитотоксических Т-лимфоцитов. Мишенью для них служат зараженные клетки, экспрессирующие на поверхности вирусные пептиды в комплексе с HLA-1. Профилактическая и лечебная эффективность этого механизма остается неясной. Лимфоциты вакцинированных животных не столь эффективны, как антитела, в предупреждении инфекции, но значительно превосходят их по протективности в ходе инфекционного процесса. Это говорит о том, что иммунологические механизмы предупреждения и выздоровления от гриппа могут иметь разные акценты в системах гуморального и клеточного иммунитета. Впрочем, согласно клиническим наблюдениям, дефекты клеточного иммунитета не оказывают заметного влияния на чувствительность к гриппозной инфекции.
В целом, рассуждая о противогриппозном иммунитете, следует помнить о формальной и фактической стороне дела. Формальная часть сводится к тому, что перенесенное заболевание (в меньшей степени бессимптомная инфекция) оставляет за собой антитела, которые сохраняются долгие годы (возможно, пожизненно) благодаря иммунологической памяти, подстегиваемой периодическим реинфицированием антигенородственными штаммами. Реальность же заключается в том, что связанный с этими антителами иммунитет имеет штаммовую направленность, не гарантируя защиты от заражения новыми дрейф-вариантами, не говоря о шифтовых субтипах. Этот парадокс ограничивает фактическую продолжительность специфической защиты до 3—4 лет (усредненное время эпидемически значимых антигенных дрейфов). Подвергаясь дрейфам (а тем более шифтам), вирус выходит из-под контроля анамнестического иммунитета, имитируя его отсутствие. Требуется много времени (точнее множество контактов с постоянно обновляющимися вирусами), чтобы иммунологический опыт стал по-настоящему действенным. На популяционном уровне это совпадает с вытеснением циркулирующего субтипа, но оставляет беззащитным против нового шифт-варианта. Каждый рецидив возбуждает новую волну иммунологической реакции, эффекторы которой притерты к обновленным антигенам вируса. Можно сказать, что иммунитет дрейфует вместе с вирусом, но этот дрейф всегда вторичен, отставая от изменчивости возбудителя. Ирония и в том, что, будучи следствием, обновленный иммунитет (антитела) тут же берет на себя роль фактора селекции иммунорезистентных клонов вируса, из которых рождаются эпидемически значимые штаммы.
И все же благодаря антигенному родству дрейф-вариантов каждая волна инфекции стимулирует прирост антител к штаммам-предшественникам, тонизируя иммунологическую память. В конце концов масштаб специфичности противогриппозных антител расширяется настолько, что способен обеспечивать защиту от близкородственных сероваров, снижая вероятность манифестных форм инфекции. Отсутствием анамнестического иммунитета объясняется повышенная чувствительность к гриппу детей, особенно младших возрастных групп.
Профилактика и терапия
В борьбе с гриппом А и В решающая роль принадлежит вакцинопрофилактике. Согласно представлениям о механизмах противогриппозного иммунитета, действие вакцинных препаратов должно быть направлено на образование антител против поверхностных антигенов, прежде всего гемагглютинина. Именно антитела против гемагглютинина являются показателем иммуногенности вакцин, а титр 1:40 говорит о надежности поствакцинального иммунитета.
Обычно для профилактики используют убитые вакцины, которые готовят из вируса, выращенного в аллантоисной полости куриных эмбрионов и инактивированного формалином или бета-пропиолактоном. Вирус подвергается очистке и доводится до нужной концентрации при помощи ультрацентрифугирования или адсорбции на макропористом стекле. Хотя некоторые из препаратов содержат цельные вирусные частицы (цельновирионные вакцины), предпочтение отдается вирусам, расщепленным на белковые субкомпоненты путем обработки эфиром и/или детергентами (субвирионные, расщепленные, или сплит-вакцины). Гемагглютинин и нейраминидазу можно подвергнуть очистке, и тогда получаются субъединичные вакцины. Разработаны и живые вакцины на основе авирулентных штаммов, в частности холодоадаптированных мутантов, размножение которых лимитировано непермиссивным порогом в 38—40оС, но хорошо происходит при нормальной температуре слизистой оболочки носоглотки (32—34оС). Их отбирают путем многократных пассажей на куриных эмбрионах при субоптимальной температуре (25оС) и дополняют (путем пересортировки) генами гемагглютинина и нейраминидазы эпидемических штаммов. Штамм, принимающий чужие протективные гены, называется вирусом-носителем, или донором аттенуации. Этот прием широко используется для получения вакцинных (не только холодоадаптированных) вирусов гриппа. Он позволяет конструировать высокопродуктивные штаммы и регулярно обновлять их актуальными антигенными вариантами.
Каждая из вакцин имеет свои достоинства и недостатки. Например, субъединичные вакцины стоят слишком дорого и требуют адъювантов для усиления иммуногенности, цельновирионные — обладают повышенной реактогенностью и применяются только для вакцинации взрослых и детей старшего возраста, живые вакцины (их вводят интраназально) менее стандартны и предсказуемы. Чаще применяют сплит-вакцины, сочетающие безвредность и высокую иммуногенность.
Но есть и общий недостаток, который обусловлен природой самого вируса и не может быть искоренен никакой технологией. Он связан с непрерывной изменчивостью вируса типа А, резко снижающей результаты вакцинопрофилактики. Чтобы избежать этого, гемагглютинин и нейраминидаза вакцинных штаммов должны быть максимально приближены к вирусам, которые могут стать причиной очередной эпидемии. Для опережающего эпидпрогноза создана глобальная сеть слежения за циркуляций вирусов гриппа. Она работает под эгидой ВОЗ, фиксируя зарождение и тенденции распространения новых дрейф- и шифт-вариантов. Это служит ориентиром для оперативного обновления вакцинных штаммов за счет пересадки генов гемагглютинина и нейраминидазы от актуальных вирусов. На подобную операцию и приготовление новой вакцины уходит около 8 мес. Учитывая скорость и масштабы гриппозных эпидемий, не всегда удается вовремя организовать массовые прививки. Приходится ограничиваться вакцинацией групп высокого риска (больные с сердечно-сосудистыми и респираторными заболеваниями, лица старше 65 лет, больные, получающие иммунодепрессанты, контингент высокого риска по эпидемическим контактам и пр.).
Вакцинация не гарантирует полной защиты от вируса, но протективный эффект на уровне популяции вполне реален: при правильном подборе штаммов заболеваемость среди привитых снижается в 2,5—3 раза. Ингредиенты вакцин отражают современную циркуляцию двух субтипов вируса гриппа А (H3N2 и H1N1) и эпидемическую значимость вируса гриппа В. Именно их актуальные (действующие на сегодня) дрейф-варианты представлены в вакцинных препаратах.
Грипп А — едва ли не единственная вирусная инфекция, которую удается предупредить при помощи химиопрепаратов. К ним относятся производные адамантана: амантадин и его улучшенный вариант — ремантадин. Механизм их антивирусного эффекта не совсем ясен, но, по-видимому, они действуют внутриклеточно, блокируя стадию позднего раздевания вириона (она связана с М2-белком), которая предшествует транспорту нуклеокапсида в клеточное ядро. Это делает понятным, почему ремантадин и амантадин влияют только на вирус А и совершенно бессильны против вируса В, лишенного М2. Впрочем, и среди штаммов типа А чувствительность сильно варьирует, вплоть до полной устойчивости к этим препаратам.
Противовирусная активность производных адамантана наиболее отчетливо проявляется при профилактическом применении: в эксперименте это спасает животных от многократной смертельной дозировки вируса. Лечебный эффект — слабее и проявляется лишь в раннем периоде заболевания. Но и профилактические схемы выглядят перегруженными. Для гарантии ремантадин требуется принимать тотчас после начала эпидемии гриппа, дважды в день по 50—100 мг, продолжая ежедневный прием до завершения эпидемической вспышки. Полагают, что таким путем можно предупредить заболевание у 70% инфицированных. Не ясно почему, но эффективность быстро снижается при запоздалом применении ремантадина, когда среди окружающих переболело более 10% человек.
Реализовать столь громоздкую схему непросто. Поэтому ремантадин показан прежде всего для людей, относящихся к группам высокого риска (см. выше). Более рациональна сочетанная химио- и вакцинопрофилактика. С этой целью в начале эпидемии делают прививку и одновременно назначают 14-дневный курс ремантадина. Этот срок необходим для формирования поствакцинального иммунитета.
Появились клинические ингибиторы нейраминидазы для ингаляционного (занамивир) и перорального (оселтамивир) применения. Установлено, что они сокращают время болезни и эффективны для профилактики гриппа А и В.
Множество работ посвящено интерферонопрофилактике гриппа. С этой целью предлагалось интраназальное закапывание или распыление лейкоцитарного (альфа) и фибробластного (бета) интерферонов. Это выглядит логичным, но интерферон, распыленный через носовые ходы, быстро выводится из респираторного тракта благодаря интенсивной секреторной деятельности эпителия и мерцанию ресничек. Если он и действует, то как краткий импульс, у которого мало шансов совпасть по времени с вирусом, запускающим инфекционный процесс. Эффекта можно ожидать лишь от многократных аппликаций больших дозировок, хотя и в этом случае достигают весьма умеренного, часто неубедительного результата. Более надежна стимуляция образования собственного (эндогенного) интерферона при помощи интерфероногенов. Количественные показатели индуцированной продукции интерферона, о которой судят по его содержанию в крови и секретах слизистых оболочек, значительно превышают возможности насыщения организма вводимыми извне препаратами. Однако и здесь требуется поддерживать активную концентрацию, т.е. пользоваться многократными инстилляциями интерфероногенов.
Подобная тактика, как и введение готового интерферона, вряд ли целесообразна при уже развившемся остром заболевании (т.е. с лечебной целью): в этом случае вполне достаточно стимулов и интерферонов, естественно возникающих в очаге поражения. Впрочем, в отечественных руководствах многократное введение интерферона и его индукторов отмечается как возможный способ борьбы с гриппозной инфекцией. Думается, что это ошибочно.
Тяжелобольным рекомендуется вводить иммуноглобулин. С этой целью применяли отечественный препарат направленного действия — противогриппозный иммуноглобулин. В конце концов его преимущества перед обычным (нормальным) иммуноглобулином оказались иллюзорными, и он был снят с производства. Но идеология, набрав инерцию, осталась: нормальный иммуноглобулин рекомендован для экстренной профилактики и лечения гриппа. Рассчитывают на действие противогриппозных антител, которые имеются в коммерческом иммуноглобулине и по логике должны создавать пассивный иммунитет против гриппа. Однако инъецируемые антитела (взрослым вводят внутримышечно 4,5—6 мл иммуноглобулина с содержанием антигемагглютининовых антител 1:400) тут же разводятся циркулирующей кровью и лимфой и, согласно элементарному расчету, не способны обеспечить защитного титра (1:40 по антигемагглютинину). Технически не разрешимой задачей является и обновление антител в соответствии с антигенными перестройками эпидемических штаммов. В коммерческих препаратах иммуноглобулина они всегда отстают от эволюции вируса, так как накопление противогриппозных антител в донорских популяциях происходит к концу эпидемии. Эти рассуждения позволяют усомниться в специфическом (вируснейтрализующем) эффекте иммуноглобулина. Его бессмыслено применять для профилактики, а полезность при тяжелых формах гриппа (в этом упорствуют некоторые клиницисты) можно связать с неспецифическим действием, которое проявляется при интоксикациях разного генеза.
Лабораторная диагностика
Диагноз гриппа нетрудно подтвердить, проследив за динамикой антител в сыворотке крови. Для этого исследуют парные сыворотки (сыворотки, полученные в ходе заболевания), отмечая нарастание антител к нуклеопротеину, гемагглютинину или нейраминидазе. Можно протестировать отделяемое носа и глотки на содержание вирусных антигенов (иммунофлюоресценция, иммуноферментный анализ) или РНК. Еще лучше выделить и идентифицировать вирус. Впрочем, необходимости в подобных исследованиях обычно не возникает. Диагноз ставят эпидемиологи, и они же следят за эволюцией инфекции и ее возбудителя. Эпиданамнез предполагает серотипирование изолированных штаммов и оценку их вклада в сероконверсию популяции, т.е. в коллективный иммунитет.
Грипп В
Вирус типа (вида) В — родственник вируса А. Они имеют 20% гомологии РНК и при смешанной инфекции дают фенотипические гибриды с образованием вирионов, имеющих сердцевину от одного вируса, а оболочку — от другого. Но генетических рекомбинаций при этом не происходит, и каждый вирус сохраняет свою дискретность.
Официального разделения вирусов гриппа В на серотипы не существует. Это связано с отсутствием четкого разграничения между штаммами по поверхностным антигенам, поскольку шифтовые варианты для вируса В не известны. Изменчивость носит более плавный характер, чем у вируса типа А. Она затрагивает главным образом гемагглютинин, и не выходит за рамки дрейфов, которые довольно мирно уживаются между собой и во время эпидемий могут циркулировать вместе. Замещение старых штаммов новыми происходит медленно.
Пандемий гриппа В не бывает. Дело ограничивается локальными вспышками, в худших случаях — эпидемиями в масштабе города или нескольких регионов одной страны. Интервалы между ними составляют 3—4 года. Наблюдаются большие (до года) промежутки, когда эпидемии не регистрируются ни в одной стране, хотя спорадическая заболеваемость не прекращается. Этому способствует непрерывная циркуляция вирусов среди населения (главным образом детей), что в принципе отличается от гриппа А, возбудитель которого после завершения эпидемии быстро перестает выделяться от людей. Это можно объяснить тем, что для вируса типа В человек — единственный естественный хозяин и неизбежность постоянных контактов между ними эволюционно закреплена.
Вспышки гриппа В могут совмещаться с гриппом А, предшествовать ему или возникать изолированно. Наиболее поражаемые группы — дети и подростки (школьники); известны эпидемии, распространяющиеся только среди детского населения. Клинически заболевание не отличимо от гриппа А, и для дифференциального диагноза требуется вирусологическое обследование. Специфическая профилактика строится на тех же принципах, что и при гриппе А.
Грипп С
Вирус гриппа С отличается от вирусов А и В по ряду фундаментальных свойств, и был момент, когда его чуть было не отлучили от семейства ортомиксовирусов. Но общих признаков оказалось больше, и с рядом оговорок родство было закреплено официально. Правда, в последних классификациях вирус С числится как отдельный род (Influenzavirus C) семейства Ortomyxoviridae. Следует отметить, что вирусы типа С не классифицируются серологически, так как у них не описано шифт-вариаций, а дрейфы, хотя и совершаются, но еще менее закономерны, чем у вируса В. В естественных условиях вирус С встречается только у человека и занимает весьма скромное место в инфекционной патологии. Он служит причиной спорадических заболеваний (чаще у детей), которые протекают как легкие и умеренно тяжелые формы гриппа, часто сопутствуют эпидемиям гриппа А и могут быть отдифференцированы от него лишь при помощи лабораторных методов. Никаких специальных мер для профилактики и лечения гриппа С не применяется.
* * * Более двадцати лет назад А.А. Смородинцев в прекрасной книге о гриппе писал: «Периодически повторяясь, грипп и ОРЗ уносят в течение всей нашей жизни около одного года. Человек проводит эти месяцы в недеятельном состоянии, страдая от лихорадки, общей разбитости, головной боли, отравления организма токсинами». Это справедливо и сегодня. Главный ущерб связан с эпидемиями гриппа А, в меньшей степени — гриппа В. Грипп С занимает скромное место среди неэпидемических респираторных вирусов, внося небольшой вклад в спорадическую заболеваемость.
Стратегия борьбы с гриппом очевидна — ее основой должна быть вакцинопрофилактика эпидемических разновидностей гриппозной инфекции. Для этого есть отличные технические возможности, которые базируются на знаниях о протективных антигенах вируса, эволюции вирусного генома, критериях антивирусной защищенности отдельных лиц и популяций. Неоднократно провозглашалось, что проблема почти решена, но результат всегда был скромнее претензий. Теперь понятно, что это не вина, а беда ученых. Мутанты и рекомбинанты вируса типа А, которые сочетают в себе антигенную новизну с высокой вирулентностью и не всегда своевременно отслеживаются эпидслужбами, создают массу трудностей, постоянно угрожая девальвацией вакцинных препаратов.
Слабость стратегии всегда рождает тактические ухищрения, часть из которых заведомо недобросовестны, а другие грешат формальной логикой и по сути являются тупиковыми. Немало подобных примеров знает и история гриппа. «Едва ли при какой-либо другой болезни было предложено и испробовано столько средств, сколько именно при инфлюэнции; но это обилие средств и доказывает, что нет ни одного верного». Об этом следует помнить, просматривая фармакологические справочники, где бесцеремонно рекламируются явно несостоятельные средства для лечения и профилактики гриппа. Что касается добросовестных заблуждений, то и они вовсе не безобидны, особенно с учетом экономической подоплеки.
Парамиксовирусы
- Вирион и структурные белки.
- Природа эпитопного консерватизма.
- Этапы репликации.
- Иммунитет и принципы этиотропной терапии.
- Вирусология и патология отдельных инфекций.
Собирательный термин миксовирусы (от греч. myxa — слизь) предложен Эндрюсом (C. Andrewes) в 1955 г. для вирусов гриппа, паротита и ньюкастлской болезни птиц. Он отражает сродство с муцинами и первоначально объединял оболочечные вирусы, способные связываться с мукополисахаридами и гликопротеинами, в том числе с сиалированными структурами клеток. Позже миксовирусы были разделены на два семейства — ортомиксовирусы (Orthomyxoviridae, вирусы гриппа) и парамиксовирусы (Paramyxoviridae), которые сильно отличаются друг от друга по биологии и строению вириона.
Парамиксовирусы человека включают вирусы парагриппа, паротита, кори и респираторно-синцитиальный вирус (РСВ). По современной классификации они входят в четыре родовых таксона:
- Paramyxovirus (вирусы парагриппа 1 и 3);
- Rubulavirus (вирус паротита, вирусы парагриппа 2 и 4);
- Morbillivirus (вирус кори)$
- Pneumovirus (респираторно-синцитиальный вирус).
Недавно появились сообщения о новом парамиксовирусе человека, который был изолирован (сначала в Нидерландах, затем в Австралии) от детей с острыми респираторными заболеваниями (ОРЗ). Он получил условное название «человеческий метапневмовирус» — hMPV (англ. human metapneumovirus). Полагают, что hMPV может быть причиной около трети ОРЗ с неустановленной этиологией.
К парамиксовирусам принадлежит также ряд вирусов млекопитающих и птиц (вирусы чумы собак и крупного рогатого скота, вирус болезни Ньюкастл птиц, возбудители ОРЗ домашних и диких животных). До недавних пор парамиксовирусы животных считались не опасными для человека. Но оказалось, что это не так. Появилось понятие зоонозные парамиксовирусы (род Henipavirus), с которыми связано несколько новых (англ. emergent) инфекций человека. Ошеломляющей явилась вспышка энцефалита в Малазии в 1998—1999 гг. Заболели 265 человек, 104 погибли. Причиной стал ранее не известный парамиксовирус, получивший название Nipah-вирус.
Заражение произошло от свиней, которые болеют Nipah-ОРЗ и гломерулонефритом, экскретируя вирус с респираторными секретами и мочой. Свиньи в свою очередь были инфицированы при контакте с одной из разновидностей летучих мышей, поддерживающих бессимптомную персистенцию Nipah-вируса. Для подавления вспышки пришлось уничтожить почти все поголовье свиней. Случаи заражения от больных людей не наблюдались.
Парамиксовирусы являются РНК-содержащими крупными оболочечными вирусами со спиральным типом симметрии. Это означает, что вирион (он имеет сферическую форму) имеет средний диаметр 150—300 нм с колебаниями от 100 до 800 нм и включает два главных компонента — нуклеокапсид, содежащий РНК-геном, и наружную липопротеиновую оболочку (суперкапсид) (рис. 1). Нуклеокапсид закручен в спираль, напоминающую винтовую лестницу, и обладает гибкостью, которая зависит от ионного состава и рН среды. Это позволяет ему скручиваться в компактную структуру (которая умещается в суперкапсид) и поддерживать плотный контакт с элементами РНК-полимеразного нуклеокапсидного комплекса.
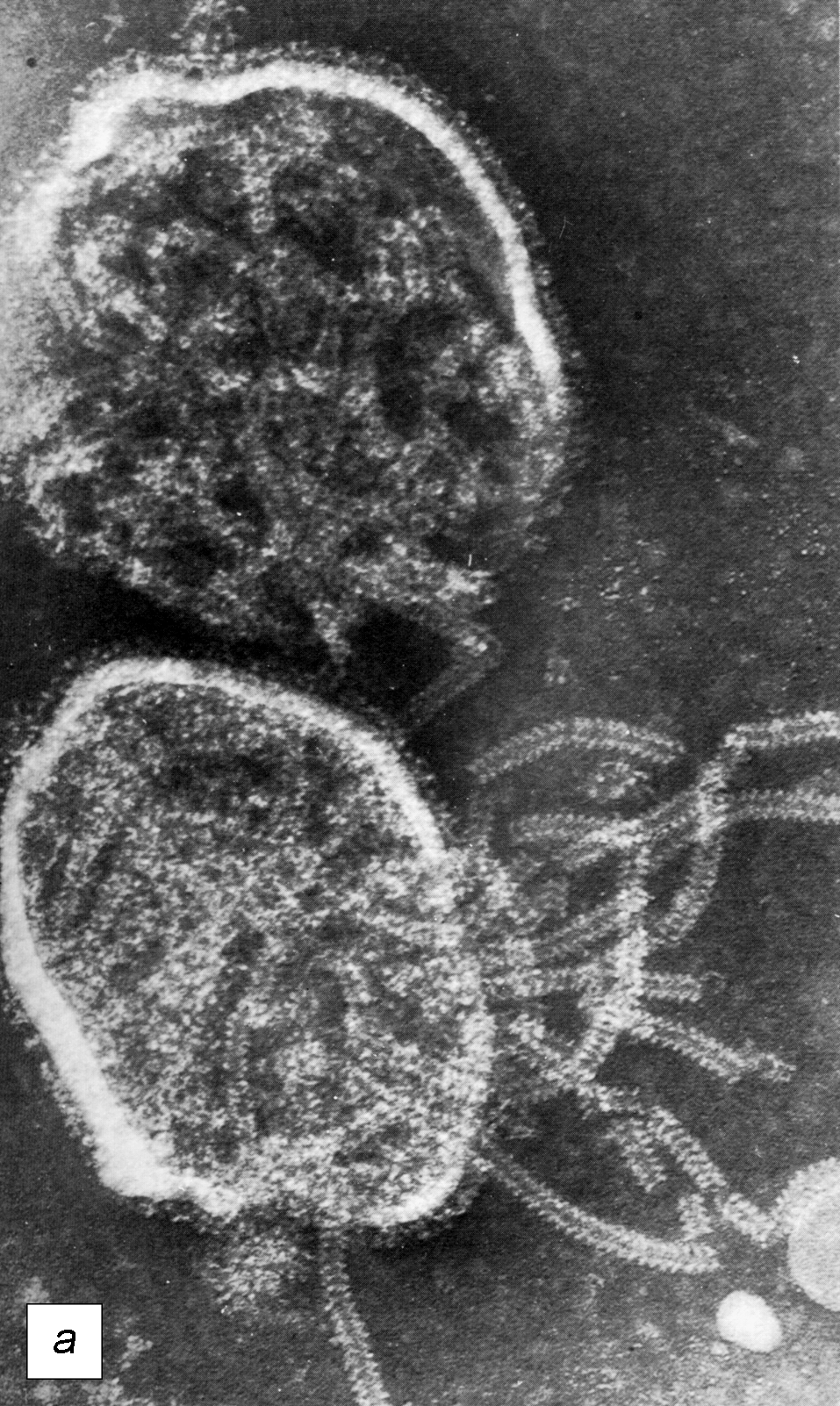
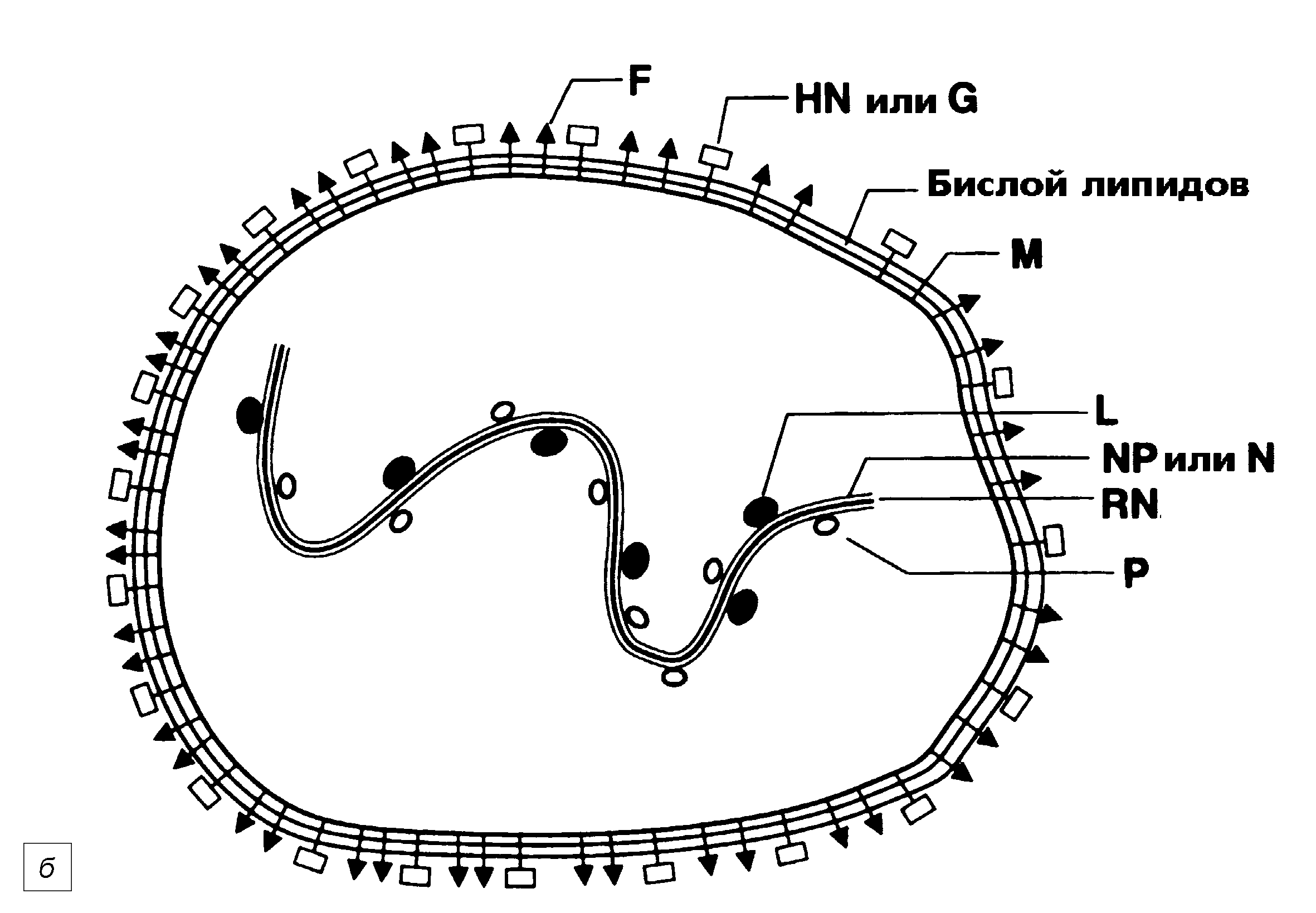


Рис. 1. Вирион парамиксовирусов: а — два вириона вируса парагриппа (электронограмма). На поверхности видна бахрома из суперкапсидных белков. Хорошо видна капсомерная (похожая на рыбий скелет) структура нуклеокапсида, частично выпавшего из нижнего вириона. Полоска равна 100 нм ; б — схема строения вириона парамиксовирусов. F — белок слияния, HN/H/G — рецепторный белок, M — матриксный белок, L и Р — белки РНК-полимеразногокомплекса, NP(N) — нуклеопротеин; RN — рибонуклеопротеин
На поверхности есть шипы, образованные двумя гликопротеинами. Более крупный наделен гемагглютинирующей и нейраминидазной активностью (вирусы парагриппа и паротита), только гемагглютинирующими свойствами (вирус кори) или лишен того и другого (РСВ). Значение этих различий не известно, но они относятся к числу классификационных признаков парамиксовирусов. Наличие или отсутствие гемагглютинирующей (Н) и нейраминидазной (N) активности отражены в акронимах — HN (вирусы парагриппа и паротита), Н (вирус кори) и G (от англ. glycoprotein; РСВ). В рецепции вирусов парагриппа и паротита участвуют сиаловые кислоты, для вируса кори главным клеточным рецептором служит CD46, на роль рецепторов для РСВ претендуют молекулы поверхностного глюкозаминоглюкана. Гемагглютинирующая и нейраминидазная активности не имеют отношения к рецепции: участки молекул, определяющие эти функции, структурно разобщены с сайтами связывания.
Второй гликопротеин, F (от англ. fusion — cлияние), формирующий более короткие шипы, вызывает слияние вирионной оболочки с мембраной клетки, обеспечивая прямое (минуя эндосому) поступление нуклеокапсида в цитоплазму. F-белок синтезируется в виде предшественника, F0, который сам по себе не активен, но обретает функцию слияния после ограниченного протеолиза. Его вызывают клеточные ферменты (фурины), относящиеся к семейству трипсиноподобных серинпротеаз. Протеолиз ведет к образованию двух субкомпонентов, F1 и F2, сохраняющих связь через дисульфидный мостик. Достаточно замены одного нуклеотида в f-гене, чтобы сделать F0 не чувствительным к протеолитической активации. Принципиально, что F0-расщепляющие ферменты в изобилии секретируются эпителиоцитами дыхательных путей. Это обеспечивает активацию F0 даже в тех случаях, когда он не успел подвергнуться внутриклеточному протеолизу. Функциональная сущность операции сводится к обнажению гидрофобного участка (пептида слияния) в составе F1, пенетрирующего клеточную мембрану.
Пептид слияния имеет структурное сходство не только у разных парамиксовирусов, но и гомологичен НА2-терминалу гемагглютинина (НА) вирусов гриппа, который выполняет такую же функцию. Но есть и принципиальное различие: F1 активен в условиях нейтральной среды и поэтому действует внеклеточно (на уровне плазматической мембраны); НА2 обретает активность после изменения конформации в кислой среде. Это происходит внутри эндосом после рецепторзависимого эндоцитоза вирионов. Только здесь (внутри клеток), внедряясь гидрофобным концом в эндосомальную мембрану, гриппозный гемагглютинин обеспечивает раздевание гриппозного нуклеокапсида и его выход в цитоплазму. Эти детали принципиальны для понимания взаимоотношений вирусов с клетками. Они служат ориентиром для разработки антивирусных препаратов, в частности структурных аналогов, блокирующих функцию пептида слияния. Сюда, казалось бы, вписываются и ингибиторы протеаз. Но сложность заключается в том, что в созревании вириона (F-белка) участвуют не вирусные ферменты, а протеазы клеток. Подавление их активности может быть небезразличным для хозяина.
Суперкапсид парамиксовирусов образуется из модифицированной плазматической мембраны. На заключительном этапе репродукции вирионы отпочковываются от клетки, прихватывая фрагменты мембраны с предварительно включенными в них гликозилированными белками HN/H/G (в зависимости от вируса — см. выше) и F. Включение в мембрану активированного F-белка определяет возможность слияния с соседними клетками с образованием поликарионов вплоть до симпласта/синцития (см. «Болезнетворность вирусов»). Это символизирует цитопатический эффект парамиксовирусов, содействуя межклеточному распространению вирусов. В этом еще одно базисное различие между пара- и ортомиксовирусами. Вирусы гриппа не способны к образованию поликарионов, так как конформационное обнажение сайта слияния в НА2-фрагменте гемагглютинина происходит внутри клеток (в кислой среде эндосом). Впрочем, синцитиальные структуры характерны главным образом для инвитровых клеточных культур. В организме симпласты (гигантские многоядерные клетки) наиболее типичны для коревой инфекции.
Между суперкапсидом и нуклеокапсидом расположен матриксный (М) белок. Он обладает сродством к поверхностным гликопротеинам и нуклеокапсидному белку (NP). Благодаря этому при почковании нуклеокапсид фиксируется на тех участках мембраны, в которые включены суперкапсидные белки, пронизывающие мембрану своими гидрофобными концами. После созревания М-белок укрепляет структурную монолитность вириона.
Нуклеокапсид содержит несегментированную РНК, ассоциированную с 3 белками (см. рисунок). Один из них, N или NP (от nucleoprotein), прочно связан с РНК и в зрелом вирионе выполняет механическую функцию, образуя капсидную оболочку, стабилизирующую вирусный геном. Два других, L и Р (от англ. large — крупный и phosphoprotein — фосфопротеин), являются элементами РНК-полимеразного комплекса.
РНК (кроме 6—7 вирионных белков в ней закодировано и несколько неструктурных белков) имеет негативную полярность, т.е. не может напрямую функционировать как мРНК. Она обретает такую способность после транскрипции, в результате которой образуются субгеномные (+)РНК. Они подвергаются трансляции, т.е. служат мРНК. Для транскрипции, а затем и репликации вирусного РНК-генома требуется РНК-зависимая РНК-полимераза. Клетки не располагают таким ферментом, и эту сугубо специфичную для (-)РНК-вирусов функцию выполняют собственные ферменты вируса. Их первая (инициирующая) порция вносится в клетку в виде РНК-полимеразного комплекса вириона. Ингибиторы РНК-зависимой РНК-полимеразы могли бы стать отличным средством для избирательного подавления вирусной репликации, но лечебные препараты такой ориентации отсутствуют.
В целом по морфологии и молекулярно-структурной организации вирионы парамиксовирусов напоминают гриппозные вирусы, но крупнее и разнороднее их. Плеоморфизм отражает вариабельность почкования и сборки вирионов, которая сопровождается их агрегацией. Это может быть связано с недостаточной активностью (или отсутствием) нейраминидазы. Склонность к агрегации может иметь функциональные последствия. Дело в том, что вирионные конгломераты мультиплоидны, так как содержат геномные молекулы нескольких вирусных клонов. Это определяет возможность генетической (и, следовательно, фенотипической) комплементации при очередном заражении клеток такого рода гибридами. Мутанты, дефектные по тем или иным признакам, помогают друг другу при репликации, замещая функции, ослабленные у другого вируса. Значение такого механизма становится понятным, если учесть, что, в отличие от вирусов гриппа, парамиксовирусы обладают слабой генетической изменчивостью, т.е. лишены механизма, который снабжал бы вирусную популяцию материалом для оперативной селекции наиболее адекватных клонов. Комплементация генных продуктов в известной мере сглаживает подобного рода генетическую инертность.
Эпитопный консерватизм поверхностных антигенов заслуживает специального комментария. Структурная монолитность несегментированного генома исключает пересортировку генов с внезапным появлением новых генных комбинаций — стратегического механизма (шифт) антигенной изменчивости вируса гриппа А. Труднее понять причину почти полного отсутствия мелких эпитопных вариаций (дрейф), которые обеспечивают непрерывное обновление суперкапсидных антигенов ортомиксовирусов. За многие годы не обнаружено существенных изменений в HN/H/G и F-белках парамиксовирусов. Этому соответствует и прочный иммунитет при системных парамиксовирусных инфекциях (корь, паротит), который может быть искусственно воспроизведен при помощи вакцинных штаммов, полученных десятки лет назад. Определенные перестройки, безусловно, имеют место, но таких изменений, которые позволили бы вирусу миновать иммунный контроль со стороны хозяина, у парамиксовирусов не замечено. Можно было бы думать, что их РНК-полимераза делает меньше ошибок, чем аналогичный фермент гриппозных вирусов, но это не так: вероятность спонтанных мутаций у пара- и ортомиксовирусов одинакова. Одним из объяснений может быть то, что большинство аминокислот жестко вписываются в структурно-функциональную канву парамиксовирусных белков и не могут быть замещены без ослабления вируса. Иными словами, мутантные клоны не получают преимуществ и поэтому не закрепляются естественным отбором. Эпитопный консерватизм поверхностных антигенов обеспечивает действенность анамнестического иммунитета, сглаживая (вирусы парагриппа и РСВ) или полностью блокируя (вирусы кори и паротита) болезнетворность вирусов при повторных контактах.
Репликативная стратегия парамиксовирусов типична для РНК-вирусов с негативным геномом. Ее реализация происходит в цитоплазме, и в этом отношении парамиксовирусы вновь отличаются от вирусов гриппа, которые действуют более изощренно, производя все подготовительные операции и конструируя новые нуклеокапсиды в ядре. Прямолинейность парамиксовирусов объясняется тем, что их РНК-полимераза (точнее РНК-полимеразный комплекс) не нуждается в экзогенных праймерах, которые могут быть заимствованы только в ядре (см. «Вирусы гриппа»). В культурах in vitro парамиксовирусы слабо влияют на клеточный метаболизм, и гибель клеток обычно происходит после образования симпласта. Это заставляет подозревать, что болезнетворность связана не только с прямым действием вирусов, но и опосредована иммунными реакциями. Подробнее мы поговорим об этом ниже.
Парамиксовирусы индуцируют образование нейтрализующих антител. Они нацелены против поверхностных белков, действуя на свободные вирионы и клетки, экспрессирующие вирусные антигены. Суперкапсидные белки — главные протективные антигены парамиксовирусов, по крайней мере на превентивном этапе. Особенно важны анти-F-антитела, которые не только подавляют инфекциозность, но и препятствуют распространению вирусов на соседние клетки, блокируя их слияние. Наиболее надежен эффект сывороточных антител при кори и паротите. Это соответствует общим принципам инвазивных инфекций, защита от которых базируется на антителах, перехватывающих вирус в циркуляции. Мукозальные антитела менее действенны, не гарантируя от реинфекций. Эффективная вакцинопрофилактика разработана только для кори и паротита. Она опять-таки основана на индукции антител, нейтрализующих вирус на этапе вирусемии.
Для полной элиминации парамиксовирусов необходимы Т-эффекторы, которые могут быть настроены не только против суперкапсидных белков, но и против внутренних компонентов вириона и даже неструктурных белков, презентируемых на поверхности зараженных клеток молекулами HLA.
Этиотропная терапия парамиксовирусных инфекций не разработана. Лечение рибавирином не принесло удовлетворения. Неэффективными оказались ингибиторы протеаз, которые применялись в надежде на подавление активации F-белка. Не меньше формальной логики было и в предложении воздействовать на вирусы препаратами нуклеаз. Эти рекомендации остались в прошлом, хотя до сих пор по инерции фигурируют в руководствах по инфекционным болезням. В качестве потенциальных мишеней для антивирусных агентов рассматриваются РНК-полимераза и F-зависимый эндоцитоз вирионов. Именно в этом направлении ведется поиск препаратов, способных остановить развитие парамиксовирусных инфекций, наиболее опасных в младенчестве (первый год жизни) и раннем детстве.
Респираторно-синцитиальный вирус
Среди изобилия возбудителей ОРЗ респираторно-синцитиальный вирус занимает особое место. РСВ-инфекция — наиболее важная причина острой патологии нижних отделов респираторного тракта детей первых двух-трех лет жизни, абсолютно лидируя среди возбудителей бронхиолита (воспаление мельчайших бронхов) и пневмонии у младенцев (первый год жизни). Максимум тяжелых заболеваний приходится на возраст от 6 нед до 6 мес с пиком в 2 мес. Такого рода возрастной профиль уникален для РСВ. Начиная с полугода вероятность бронхиолита снижается, но в этиологии пневмоний лидерство РСВ сохраняется до 5 лет. При повторных заражениях РСВ теряет агрессивность: инфекция проявляется как малая простуда. Это типично для детей старшего возраста и, тем более, взрослых.
Подобно другим ОРВИ-вирусам, РСВ убиквитарен, и всегда есть люди, которые им инфицированы. Вопреки бытующему мнению, аэрогенный путь — не главный способ распространения РСВ, и медицинские маски не спасают от заражения при внутригоспитальных вспышках. Чаще вирус передается через руки и предметы обихода, загрязненные инфицированными мукозальными секретами, при аппликации на слизистую оболочку носовой полости или конъюнктиву. Для этого необходимы тесные контакты, так как на объектах внешней среды РСВ выживает не более 6 ч.
Согласно сероэпидемиологическим данным (наличие анти-РСВ-антител), около половины детей инфицируются на первом году жизни, а к двухлетнему возрасту конфликт с вирусом имеют более 90%. Первичная инфекция почти всегда клинически значима, начинаясь как малая простуда, которая в 10—40% случаев осложняется поражением нижних отделов дыхательного тракта. Тяжелые формы, требующие госпитализации, наблюдаются у 1—3% детей. С учетом массовости заражения это формально небольшое число достигает внушительных размеров в абсолютном исчислении. Характерны ежегодные эпидемические вспышки. Их пик обычно приходится на один из трех зимних месяцев, когда число младенцев с бронхиолитом и пневмонией коррелирует с распространением РСВ. Любой необычно высокий подъем такой патологии следует воспринимать как РСВ-инфекцию. РСВ замечен и как причина нозокомиальных инфекций в яслях и детских больницах. У персонала и родителей, контактирующих с больными детьми, часто возникают простудные заболевания. РСВ зарегистрирован и как оппортунистический патоген среди иммунодефицитных лиц и пожилых людей, в частности в домах престарелых.
РСВ (первые штаммы изолированы в 1956—1957 гг.) принадлежит к роду Pneumovirus, куда, кроме него, входят вирус пневмонии мышей, РСВ коров, овец и коз. Эпитеты «респираторный» и «синцитиальный» не определяют специфики вируса, так как тропизмом к респираторному эпителию и способностью индуцировать образование симпласта-синцития обладают все парамиксовирусы. Название связано с тем, что РСВ был одним из первых вирусов, в клеточных культурах которого были отчетливо зафиксированы фигуры синцития.
В отличие от других парамиксовирусов поверхностные РСВ-белки не агглютинируют эритроциты и, подобно вирусу кори, лишены нейраминидазной активности. Однако это не отражается на взаимоотношениях с клетками, которые складываются так же, как у остальных парамиксовирусов. Просто вместо гемагглютинина (H/HG) рецепторную функцию у РСВ выполняет суперкапсидный гликопротеин G.
Геном РСВ устроен сложнее, чем у других парамиксовирусов, кодируя несколько дополнительных белков, среди которых два неструктурных белка с неизвестными функциями (NS1 и NS2), М2-белок, входящий в РНК-полимеразный комплекс вириона, и трансмембранный SH-гликопротеин (он есть также у вирусов парагриппа 2 и 4). SH-белок участвует в слиянии вирионной и клеточной мембран, но это вряд ли имеет существенное значение, так как анти-SH-антитела лишены вируснейтрализующих свойств. Протективными антигенами являются G- и (особенно) F-белок: их блокада антителами подавляет развитие инфекции.
По антигенным особенностям G-белка предложено дифференцировать две группы РСВ — А и В. Штаммы А (полагают, что они вызывают более тяжелые заболевания) и В циркулируют одновременно, хотя их лидерство периодически меняется. Внутри групп вирусы тоже неоднородны. Это может быть одной из причин ненадежности анти-РСВ-иммунитета и его постепенного усиления под влиянием повторных инфекций.
Вирус первично размножается в назальном эпителии, откуда аспирируется в нижние отделы дыхательного тракта. Не исключено, что диссеминация происходит и через межклеточные контакты при образовании синцития. В этом случае вирус не выходит из клеток, скрываясь от антител. РСВ размножается и в макрофагах, хотя гораздо медленнее, чем в эпителиоцитах. Полагают, что макрофаги участвуют в распространении инфекции, содействуя развитию интерсцитиальной пневмонии. Иногда РСВ достигает среднего уха, вызывая средний отит. Дальше этого дело не идет, вирусемия для РСВ большая редкость. РСВ удавалось выделить из других органов (почки, миокард, печень), но это были фатальные случаи на фоне глубоких дефектов клеточного иммунитета.
Инкубационный период не превышает 5 дней, т.е. вполне типичен для инфекций, когда входные ворота и патогенетически значимые мишени совпадают. У младенцев симптомы поражения нижних отделов респираторного тракта (одышка, свистящее дыхание, хрипы) возникают через 1—3 дня после ринореи, отражая распространение процесса на бронхи и бронхиолы. Вирус продолжает здесь размножаться, но не пенетрирует эпителия. Выделение РСВ с респираторными секретами затягивается на 1—3 нед, часто продолжаясь после клинического выздоровления.
Патологические изменения аналогичны бронхиолиту и пневмонии, вызываемым другими респираторными вирусами, такими как вирусы гриппа, парагриппа и аденовирусы. При бронхиолите происходит разрушение реснитчатых эпителиоцитов (в первичных культурах респираторного эпителия РСВ быстро вызывает цилиостаз, что, по-видимому, имеет отношение к патогенезу РСВ-инфекции), в тяжелых случаях наблюдается массивный некроз эпителия с перибронхиальными мононуклеарными инфильтратами, отеком подслизистой и бронхореей. Это ведет к обструкции бронхиол с очагами ателектаза и компенсаторной эмфиземы. Альвеолы могут быть заполнены воспалительным экссудатом. Образование синцитиальных пластов из эпителиоцитов in vivo не доказано, но слущивание погибающих клеток способствует закупорке бронхиол. Развитию обтурационного синдрома у детей содействует и минимальный просвет бронхиол, теряющих проходимость даже при умеренном повреждении и воспалении стенки. Этим объясняется критический характер болезни в младенчестве.
Человек высокочувствителен к РСВ, и только иммунитет, обретаемый с годами (при повторных контактах с вирусом) гарантирует защиту, по крайней мере от тяжелой инфекции. Как и при других ОРВИ, эффекторы иммунитета включают мукозальные IgA-антитела, сывороточные антитела и Т-лимфоциты. Каждый из этих механизмов имеет свои особенности, внося конкретный вклад в антивирусную защиту. Вируснейтрализующей активностью обладают только антитела против поверхностных антигенов вириона (G и F), причем не все их эпитопы наделены протективностью. IgA-антитела препятствуют заражению эпителия верхних дыхательных путей, но не спасают от поражения нижних отделов респираторного тракта, т.е. от наиболее тяжелых форм заболевания. С этой задачей лучше справляются сывороточные IgG-антитела, проникающие в мукозальные секреты бронхов и альвеолы. На этом основано профилактическое применение внутривенного анти-РСВ-иммуноглобулина в периоды эпидемической опасности, прежде всего у младенцев из групп повышенного риска.
С динамикой мукозальных и сывороточных антител отчасти связаны возрастные особенности РСВ-инфекции и вероятность повторных заболеваний. Суть сводится к следующему. Практически все новорожденные получают сывороточные IgG-антитела от матери. При грудном вскармливании к ним добавляются антитела, поступающие с молоком. Однако не всегда и те и другие достигают протективных титров. Более того, в результате естественного катаболизма содержание материнских антител ежемесячно падает на 50%. К 2—4 мес их уровень обычно становится ниже протективного, а к 7 мес все антитела являются результатом активного ответа на инфекцию. Именно они определяют устойчивость к РСВ-бронхиолиту и/или пневмонии. Но многие младенцы слабо реагируют на РСВ-антигены, что объясняется их иммунологической незрелостью и супрессивным действием материнских антител. Это относится как к сывороточным, так и к секреторным антителам, определяя низкий уровень противовирусной защиты. Со временем ситуация выравнивается, но относительно короткая память мукозального IgA-иммунитета и серологическая неоднородность вируса предрасполагают к повторным РСВ-поражениям верхних дыхательных путей. Нижние отделы защищены более надежно. Это связано с непростым маршрутом, который вирус должен пройти до терминальных бронхов и альвеол, а также с относительной стабильностью защищающих их сывороточных IgG-антител. Вкупе с физическими особенностями взрослого респираторного тракта (более широкие воздухоносные пути) это практически исключает вероятность клинически значимых бронхиолита и пневмонии. Повышенная опасность сохраняется лишь для групп риска — кардиопульмонологические и иммунодефицитные больные, старые ослабленные люди.
Предупреждая и/или смягчая течение инфекции, антитела менее активны в элиминации вируса. По крайней мере попытки применения анти-РСВ-иммуноглобулина с лечебной целью дали противоречивые результаты и в целом не принесли удовлетворения. Возможно, это связано с тем, что клинические симптомы возникают после того, как вирус прочно обосновался в клетках респираторного эпителия и малодоступен для эффекторов гуморального иммунитета. Складывается впечатление, что для скорейшего уничтожения вируса более важны анти-РСВ Т-лимфоциты, которые убивают вирусинфицированные клетки и вместе с ними удаляют вирус. У детей с нормальной иммунной системой исчезновение вируса из респираторных секретов происходит в среднем через 20 дней, при нарушениях клеточного иммунитета — через несколько месяцев.
В отличие от гриппа и парагриппа РСВ-инфекция обычно не вызывает повышения интраназального уровня интерферона, и скорее всего он не участвует в выздоровлении.
Неожиданный поворот в иммунологических представлениях РСВ-инфекции произошел после испытаний в 1960-х гг. убитой РСВ-вакцины. Несмотря на то, что вакцинация вызывала образование антител, дети продолжали болеть. Более того, чаще, чем у непривитых, инфекция протекала в тяжелой форме. Возникли подозрения, что иммунологические механизмы не только не защищают от РСВ-инфекции, но и включаются в ее патогенез. Было установлено, что иммунный ответ при парентеральном введении убитой вакцины отличается от того, что наблюдается при естественной РСВ-инфекции. Такая вакцинация не обеспечивает образования достаточного количества мукозальных IgA-антител и поэтому малоэффективна. Неважно обстоит дело и с сывороточными антителами. Несмотря на формальное изобилие, они обладают слабой вируснейтрализующей активностью, возможно, из-за повреждения протективных эпитопов G- и F-белков при инактивации вируса. Еще больше тонкостей обнаружено при изучении Т-клеточного иммунитета. В отличие от естественной инфекции при вакцинации преобладают реакции Th2-клеток, которые, с одной стороны, малоэффективны против вирусов (низкая наработка цитотоксических Т-лимфоцитов), а с другой — предрасполагают к развитию иммунологического воспаления в стенке бронхиол. Именно это скорее всего служит основой обтурационного синдрома, клинически сходного с приступами бронхиальной астмы. Многочисленные исследования показали, что РСВ-инфекция действительно сопровождается признаками аллергического (IgE-зависимого) воспаления, а первичная болезнетворность РСВ усиливается механизмами, которые призваны ее подавить. В этом диалектика («добро» и «зло») иммунитета и воспаления. Здесь «зло» особенно заметно на фоне легко наступающей обструкции бронхиол у младенцев.
Неудача первого опыта вакцинации не остановила поисков, но исследования стали менее прямолинейными, если не сказать осторожными. Высокая заболеваемость и даже смертность среди детей от бронхиолита сохраняет за РСВ позицию одного из первых кандидатов на создание вакцины. Она должна быть безопасной и эффективной уже в младенчестве, т.е. для детей первых месяцев жизни. В этом основная сложность, если учесть иммунологическую незрелость новорожденных и торможение иммунного ответа антителами, пассивно получаемыми от матери. Следует учитывать и антигенную неоднородность двух основных (А и В) групп вируса, которая может потребовать усложнения препарата. Меньше проблем ожидается от внутригрупповой гетерогенности G-белка. Ведется разработка вакцин на основе рекомбинантного F-белка и живых температурочувствительных штаммов, аттенуированных при многократных холодовых пассажах.
Сомнения по поводу возможности создания эффективной вакцины связаны с тем, что РСВ-инфекция нередко возникает на фоне, казалось бы, достаточно выраженного иммунитета (например, в присутствии вируснейтрализующих антител), а повторные заболевания могут возникать при заражении идентичными штаммами. Отметим и то, что убитые парентеральные вакцины не всегда обеспечивают надежную защиту слизистых оболочек. Логичнее выглядит введение антигенов (лучше живых аттенуированных штаммов) непосредственно в респираторный тракт — аэрозольно или интраназально.
Вирусы парагриппа
Вирусы парагриппа были изолированы в 1955—1958 гг. из носоглоточных смывов у детей с гриппоподобными заболеваниями. Это несколько родственных парамиксовирусов, которые различаются по антигенным особенностям поверхностного гликопротеина (HN) и вызывают острые респираторные заболевания у детей, реже у взрослых. Известно 4 серотипа, один из которых (серотип 4) включает два подтипа, 4А и 4В; некоторые авторы считают подтип 4В самостоятельным, пятым серотипом. К парагриппозным вирусам человека примыкают вирусы, поражающие респираторный тракт птиц (болезнь Ньюкасл), грызунов (Сендай), рогатого скота и приматов.
До недавних пор вирусы парагриппа вместе с вирусом паротита относили к роду Paramyxovirus. По новой классификации вирусы парагриппа 2, 4 и вирус паротита выделены в самостоятельный род, Rubulavirus; вирусы типов 1 и 3 закреплены за родом Paramyxovirus. Однако на практике о парагриппозных вирусах по-прежнему говорят как о единой группе, стремясь подчеркнуть клинико-эпидемиологические особенности вызываемых ими инфекций. Сделать это непросто, и лишь внимательный вирусологический анализ позволяет отдифференцировать различные варианты парагриппозной инфекции от ОРВИ иной этиологии. И все-таки есть тенденции, которые характерны для вирусов парагриппа, по крайней мере для некоторых из них.
Заражение происходит контактным путем (предметы, инфицированные респираторными секретами) и через крупно-дисперсные аэрозоли. Репликация ограничена скорее всего респираторным эпителием, вирусемия не типична. Инфекция может быть бессимптомной или ограничиться слизистой оболочкой носа и глотки, но иногда захватывает нижележащие отделы респираторного тракта. По числу случаев бронхиолита и пневмонии на первом году жизни вирусы парагриппа уступают только респираторно-синцитиальному вирусу, а в качестве причины ларинготрахеита (крупа) лидируют среди других ОРВИ-вирусов. Так же, как РСВ, парагриппозные вирусы наиболее опасны для младших детей. С возрастом чувствительность к ним падает, и клинические проявления, как правило, ограничиваются банальной простудой. Сказывается иммунитет, приобретаемый в течение жизни, хотя он и не абсолютен.
Патогенетический потенциал разных серотипов неодинаков. Наиболее серьезные проблемы связаны с серотипами 1—3, серотип 4 минимально агрессивен. Причины этого не ясны. Одним из факторов может быть неодинаковая чувствительность вирусов к протеазам респираторного тракта, которые активируют суперкапсидный белок слияния (F0), обеспечивая продолжение инфекции (см. выше). Впрочем, это не доказано.
Согласно сероэпидемиологическим данным статистика взаимоотношений с вирусами парагриппа выглядит следующим образом. Примерно 75% детей первых пяти лет жизни уже были в иммунном конфликте с серотипами 1 и 2. Для вируса типа 3 этот показатель еще выше: антитела к этому вирусу определяются у 50% детей первого года жизни и у 95% шестилетних детей. Около половины детей до пяти лет инфицируются типами 1 и 2 бессимптомно, большинство остальных переносят инфекцию в легкой форме. Всего у 3% зараженных возникает опасный круп (ларинготрахеит с признаками асфиксии), но именно это формально небольшое число определяет лидерство вирусов парагриппа, прежде всего типа 1, в развитии данного заболевания. Круп чаще регистрируется во время осенних и летних вспышек, которые повторяются раз в два года, отражая ослабление иммунной прослойки среди детского населения. Основные проблемы возникают с 4-месячного возраста, так как к этому моменту ребенок лишается иммунитета, который он получает за счет материнских антител.
Существует предположение, что развитию крупа способствует гиперпродукция вирусспецифических IgE-антител, которая предрасполагает к дегрануляции тучных клеток и зависит от генетического фона инфицируемого организма. Этому соответствуют наблюдения о более тяжелых инфекциях у лиц с аллергическими заболеваниями, что нередко истолковывается весьма расплывчато, как отражение «общей гиперреактивности респираторного тракта». Но может быть и наоборот: ОРВИ-вирусы (в том числе вирусы парагриппа) извращают чувствительность респираторного эпителия к другим раздражителям, содействуя формированию того, что принято называть аллергическим фоном (см. выше). Тяжелому течению ларинготрахеита у младших детей способствует небольшой просвет воздухоносных путей, легко закупориваемых даже при небольшом воспалительном отеке, тем более что жесткие (хрящевые) кольца трахеи препятствуют ее расширению. У взрослых дело ограничивается осиплостью (поражение голосовых связок).
Инфицирование вирусом типа 2 эпидемиологически и клинически мало чем отличается от заражения вирусом типа 1, но протекает мягче и реже сопровождается крупом. Напротив, вирус типа 3 ведет себя явно иначе. Он чаще, чем другие парагриппозные вирусы, вызывает поражения нижних отделов респираторного тракта, являсь второй по значимости причиной (после РСВ) бронхиолита и пневмонии у детей первого полугода жизни. Складывается впечатление, что материнские антитела в этом случае не оказывают должной поддержки. Вирус высококонтагиозен и вирулентен, обнаруживая выраженную эндемичность для детских лечебных и воспитательных учреждений. Эпидемические вспышки не типичны, инфекция не имеет явной сезонности, наблюдаясь на протяжении всего года. Около 75% детей к 4-летнему возрасту переносят инфекцию в клинически значимой форме. Обычно это простуда, но у трети зараженных заболевание осложняется бронхитом и/или пневмонией. Спорадические случаи наблюдаются и в дальнейшем (старшие дети, взрослые), но они протекают гораздо мягче, чем инфекции раннего детства.
Вирус типа 4 не вызывает серьезной патологии даже при первичном инфицировании детей.
Вирус эпидемического паротита (свинки)
Вирус (вместе с вирусами парагриппа 2 и 4) относится к роду Rubulivirus и по своим вирусологическим свойствам мало чем отличается от других парамиксовирусов. Но его медицинская экология и связанные с ней патогенетические ресурсы весьма своеобразны. Вирус вызывает у человека специфическую (т.е. диагностируемую клинически) инфекцию, которую собирательно называют свинкой или эпидемическим паротитом. Дело в том, что основным признаком заболевания является двухстороннее или односторонее увеличение околоушных слюнных желез (паротит), возможно, из-за воспалительной реакции в слюнных протоках, создающей препятствие для выделения слюны. Но болезнь этим не ограничивается. Инфекция носит генерализованный характер, так как вирус проникает в кровь и реплицируется в эпителиальных клетках внутренних органов. Обычно это не имеет серьезных последствий, так как, напрягая иммунитетные ресурсы, хозяин своевременно уничтожает вирус. Но возможны и осложнения, реальная значимость которых в абсолютном исчислении определяется степенью распространения инфекции. Патология определяется не только прямой агрессивностью вируса (в клеточных культурах цитопатический эффект невелик), но и отражает вмешательство иммунологически зависимых реакций.
Вирус инфицирует только людей, других природных резервуаров (как и для большинства остальных парамиксовирусов человека) не существует. В эксперименте паротит удается воспроизвести у обезьян при введении вируссодержащего материала (например, слюны больного) в проток Стенсена околоушной железы. Именно таким путем в 1934 г. был выделен первый штамм вируса.
В довакцинальный период свинка была одной из самых распространенных детских инфекций. Впрочем, и сегодня она встречается повсеместно в виде спорадических случаев или вспышек, особенно среди уплотненных коллективов (отсюда эпитет «эпидемический»). Максимум заболеваемости падает на возраст 5—15 лет. До 5 лет заболевание протекает атипично, в виде простуды. Но и в дальнейшем около трети детей переносят инфекцию субклинически или в атипичной (недиагностируемой) форме. Однако это не делает их незаразными, так как вирус выделяется со слюной и респираторными секретами. К факторам, которые во многом обесценивают противоэпидемические мероприятия, следует добавить расплывчатость инкубационного периода (7—25 дней, чаще 2—3 нед), а также то, что эпидемически значимое выделение вируса из организма начинается за 5—6 дней до клинических симптомов. Это характерно для всех инвазивных вирусных инфекций.
Экскреция вируса прекращается на 5—6-й день после развития паротита, и с этого момента больной не опасен для окружающих. Новорожденные защищены материнскими антителами, по крайней мере в течение 6 мес. В этом есть логика: вирус обязан пройти через этап вирусемии, когда он особенно уязвим для антител. Впрочем, не все так просто. Придерживаясь такого объяснения, трудно, например, понять, почему антитела не обладают профилактическим эффектом и не предупреждают развития осложнений даже при немедленном введении.
В целом свинка менее заразна, чем корь или ветряная оспа. Поэтому прослойка лиц, избежавших инфицирования, здесь больше, что оставляет шансы заболеть в подростковом и зрелом возрасте. Есть наблюдения, что серопозитивность наблюдается у 60% взрослых людей (для кори — 90%), хотя по другим данным антитела к вирусу паротита имеют около 90% восьмилетних детей. Устойчивость к инфекции лучше всего коррелирует с антителами против HN-гликопротеина (у вируса паротита его иногда называют V-антигеном). Они накапливаются медленно, через 4 нед после первых признаков инфекции, но сохраняются многие годы.
Патогенез сводится к следующему. Вирус, который содержится в слюне и респираторных секретах, распространяется воздушно-капельным путем или через инфицированные предметы. Ранее полагали, что он через проток Стенсена тотчас попадает в околоушную железу, начиная отсюда инвазию. Но сегодня большинство склоняется к тому, что репликация вируса начинается в эпителии верхнего дыхательного тракта и в регионарных (шейных) лимфоузлах. Только после этого, попав в кровь (первичная вирусемия), вирус инфицирует слюнные железы. Этому соответствует многодневная инкубация, предшествующая паротиту. Она отражает сложность маршрута, который обязан пройти вирус от входных ворот инфекции до патогенетически значимых мишеней. Считают, что именно в слюнных железах (кроме околоушных поражаются подъязычная и подчелюстная железы) вирус находит оптимальные условия для репликации и, накопившись здесь, вновь, но уже в гораздо большем количестве, проникает в кровь (вторичная вирусемия), получая возможность для диссеминации в другие ткани.
Паротит, которому предшествуют и сопутствуют симптомы общей интоксикации, является самым манифестным и постоянным (95%) признаком болезни, сохраняясь 7—10 дней. Другие проявления возникают через 4—10 дней от начала паротита и рассматриваются как осложнения, связанные с агрессивностью вторичной вирусемии. Чаще всего страдает центральная нервная система — асептический менингит или менингоэнцефалит. Они протекают доброкачественно и, как правило, не оставляют последствий. Более опасны (бесплодие!) поражения яичек и яичника, особенно в постпубертатном периоде. После 13 лет орхит и оофорит развиваются в 20% случаев, но редко ведут к стерилизации, так как воспаление обычно ограничено одной железой. Очаги поражения изредко возникают и в других органах (поджелудочная железа, суставы, миокард, слезные железы), давая соответствующую симптоматику. Вирус часто инфицирует почки и выделяется с мочой, но признаки нефрита обычно отсутствуют.
Иммунитет — пожизненный, возможно, за счет периодического реинфицирования. Протективность связана с сывороточными антителами (анти-HG, анти-F), нейтрализующими вирус на этапе первичной вирусемии. Тем более удивительно, что препараты иммуноглобулина, содержащие те же антитела, не предохраняют от заболевания и не снижают вероятности осложнений. По крайней мере, здесь далеко до позитивного единодушия, которым пользуется противокоревой иммуноглобулин (см. ниже). Может быть, не следует отрицать вероятности ранней (на этапе первичной вирусемии) генерализации инфекции с проникновением вируса не только в слюнные железы, но и в другие ткани, где, укрываясь от антител, он получает условия для медленной, но стратегически важной репликации. Другим объяснением может быть участие в протективных реакциях Т-лимфоцитов, тем более что в свое время немало дискуссий было посвящено корреляциям между замедленной гиперчувствительностью к вирусным антигенам (кожные тесты) и резистентностью к паротитной инфекции.
Создание эффективной вакцины на основе живого аттенуированного штамма вируса паротита во многом охладило споры вокруг шероховатостей и противоречий, связанных с патогенезом и иммунологией свинки. Вакцину вводят подкожно детям старше 15 мес, а также подросткам и взрослым, не болевшим паротитом. Об ее иммуногенности свидетельствуют вируснейтрализующие антитела, которые появляются у 95% привитых и сохраняются по крайней мере 10 лет.
Вирус кори
Корь остается одной из самых опасных инфекций. Ежегодно в мире болеют 20—30 млн. детей, из которых не менее 1 млн. погибают. Большинство случаев приходится на экономически отсталые страны, где до сих пор не налажена масштабная вакцинация. Это резервуар, откуда вирус угрожает популяциям с хорошо поставленной специфической профилактикой.
Особенности вируса. Вирус кори относится к роду Morbillivirus, который включает наиболее агрессивные парамиксовирусы. Кроме вируса кори он объединяет возбудителей чумы собак (поражает широкий круг плотоядных), крупного рогатого скота, овец и коз (опасен также для свиней), морских млекопитающих (китов, дельфинов, тюленей), смертельной пневмонии лошадей (известны случаи фатального заражения людей). Первые два связаны с вирусом кори антигенным родством и не дают перекрестных реакций с другими парамиксовирусами. Они вызывают сходные заболевания, нередко с фатальным исходом. Достаточно вспомнить о собачьей чуме (англ. canine distemper), особенно опасной для щенков; она протекает с тяжелым диарейным синдромом, поражением центральной нервной системы и легких, убивая или оставляя тяжелые последствия.
Вирус кори не только крайне контагиозен, но и высоковирулентен: в отличие от других парамиксовирусов бессимптомного заражения практически не бывает. При внесении вируса в популяцию, лишенную эндемичной инфекции (она поддерживает коллективный иммунитет), эпидемия неизбежна, причем болеет почти 100% населения. Последствия подобных вспышек (они известны из истории освоения европейцами новых земель, прежде всего Америки) бывали катастрофичны. Яркий пример — вспышка кори, случившаяся в 1846 г. среди жителей Фарерских Островов после общения с иммигрантами из Европы. В течение 6 нед переболело около 4000 человек. Исключение составили всего 5 человек старше 60 лет, которые много лет назад пережили последнюю коревую эпидемию и благодаря этому приобрели иммунитет.
Вирус кори похож на другие парамиксовирусы. Тем не менее есть различия, которые, возможно, имеют отношение к патогенезу инфекции:
- суперкапсидные шипы содержат только гемагглютинин, но не обладают нейраминидазной активностью (это же характерно для респираторно-синцитиального вируса);
- спектр гемагглютинационной активности ограничен эритроцитами обезьян; вирус кори не агглютинирует эритроциты человека, кур, морских свинок, мышей и пр., которые агглютинируются другими парамиксовирусами;
- связывание вириона не зависит от сиаловых кислот, но основано на взаимодействии с CD46 и, возможно, с другими мембранными структурами;
- цитопатический эффект сопровождается формированием не только цитоплазматических, но и внутриядерных включений (хотя репликация, как и у других парамиксовирусов, происходит в цитоплазме);
- симпластообразующая активность проявляется не только in vitro, но и в тканях инфицированного организма;
- в перевиваемых клеточных культурах кроме острой может быть смоделирована и персистентная инфекция без продукции инфекционного вируса и цитопатогенного эффекта;
- кроме эпителиальных клеток вирус размножается в мононуклеарных фагоцитах, лимфоцитах и эндотелиальных клетках.
Вирус кори имеет единственный серотип, не претерпевший существенных изменений с момента открытия (первый штамм изолирован J.F. Enders и T.C. Peebles в 1954 г.). Эпитопные различия, регистрируемые между штаммами при помощи моноклональных антител, не влияют на антигенную монолитность вируса в реакциях протективного иммунитета. Единственным природным резервуаром вируса является человек, хотя искусственно могут быть инфицированы обезьяны, собаки, мыши и другие животные. Полноценную картину кори удается воспроизвести только на обезьянах.
Патогенез. Основной поражаемый контингент — дети 5—6 лет, но вакцинация изменила эпидемиологические параметры, в том числе возрастные приоритеты, сместив их на школьный возраст. Заражение обычно происходит воздушно-капельным путем. В течение нескольких дней вирус размножается в эпителиальных клетках дыхательного тракта, в макрофагах и дендритных клетках регионарных лимфатических узлов, откуда распространяется (первичная вирусемия) во все отделы лимфоидной и ретикуло-эндотелиальной ткани. Здесь продолжается его размножение, после чего следует вторая, более мощная волна вирусемии инфекции. Вирус переносится лейкоцитами, прежде всего моноцитами. Удивительно, но его не удается обнаружить в лимфоцитах. Возможно, инфицированные лимфоциты быстро удаляются из циркуляции, погибая в лимфоидной ткани, или просто не поддерживают репродукцию вируса.
Вирус проникает практически во все органы. Это объясняется заражением таких убиквитарных клеток, как макрофаги, эпителиоциты кожи и слизистых оболочек. Но центральным событием, безусловно, является инфекция эндотелиальных клеток мелких сосудов. Именно в зоне их поражения формируются воспалительные реакции, которые вносят решающий вклад в развитие болезни. Многоядерные гигантские клетки с внутриядерными включениями можно обнаружить в лимфоидной ткани (лимфатические узлы, селезенка, миндалины, аппендикс), эпидермисе, конъюнктиве, печени, почках, респираторном тракте, кишечнике. Инкубация составляет 9—14 дней, хотя иногда затягивается до трех недель. Продромальный период (его называют катаральным) совпадает с вторичной вирусемией и агрессивной диссеминацией вируса. Он продолжается 2—4 дня, напоминая ОРЗ (ринит, фарингит, кашель, конъюнктивит, умеренные признаки интоксикации). Уникальный признак — энантема («внутренняя сыпь») на слизистой оболочке щек. Это так называемые пятна Коплика—Филатова — очаги некроза с зоной воспалительной гиперемии диаметром 1—3 мм. Их появление позволяет поставить диагноз до «настоящей кори».
«Настоящую» корь (она следует за продромальным периодом) знаменуют папулезная, часто сливающаяся сыпь (экзантема) с характерной динамикой (начинаясь с шеи и лица, она распространяется на тело и конечности) и тяжелая интоксикация, прежде всего высокая температура. Экзантема представляет собой очаги воспаления эпителиальной ткани, которые формируются на основе иммунологически зависимого повреждения вирусинфицированного эндотелия мелких сосудов. У больных с дефектами клеточного иммунитета сыпь отсутствует, хотя другие проявления коревого синдрома протекают не менее тяжело и часто более опасны. На пике экзантемы состояние выглядит критическим, но через несколько дней обычно все заканчивается благополучно. Осложнения возникают редко. Чаще всего (5—15%) это вторичные бактериальные инфекции — средний отит, синуситы, бронхопневмония. К ним предрасполагает нарушение клиринговой функции респираторного тракта (повреждение мерцательного эпителия) и временное ослабление иммунитета, типичное для коревой инфекции. Следствием прямой вирусной агрессии может быть интерстициальная пневмония, для которой характерны множественные очаги симпластообразования в легких (рис. 2). Такого рода гигантоклеточная пневмония развивается на фоне иммунодефицитных состояний у детей в результате неконтролируемого размножения вируса и обычно фатальна.

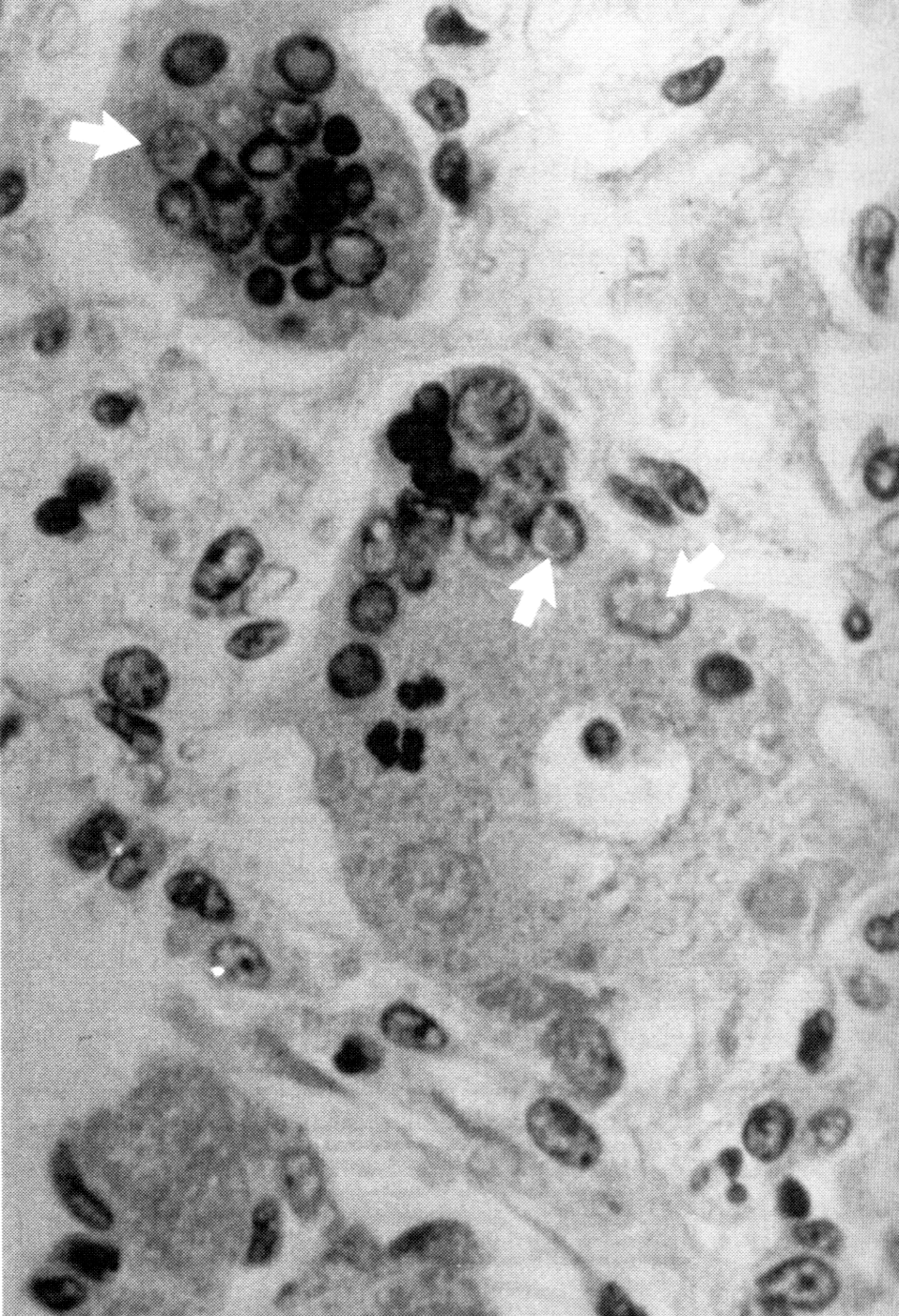
Нередко возникает диарея — следствие вирусиндуцированного поражения энтероцитов. Но опаснее всего поражение центральной нервной системы. По данным электроэнцефалографии вовлечение ЦНС в инфекционный процесс наблюдается у половины детей, но, как правило, заканчивается благополучно. Лишь в одном из 1000—2000 случаев развивается острый коревой энцефалит. Он смертелен для 5—30% детей; у 20—40% выживших остаются тяжелые последствия (глухота, умственная отсталость, приступы судорог). Еще более драматичным является то, что энцефалит возникает на фоне выздоровления (чаще через 6 дней после начала сыпи) и не связан с тяжестью коревого синдрома. Инфекционный вирус редко выделяется из головного мозга, что позволяет думать об иммунологически опосредованной патологии, подобно поражению кожи и слизистых оболочек.
Неврологические осложнения этим не исчерпываются. Очень редким (1:300000—1:1000000), но символичным для вирусной персистенции заболеванием является подострый склерозирующий панэнцефалит (ПСПЭ). Он развивается через 5—15 лет после коревой инфекции, прогрессирует в течение 6—9 мес (иногда трех лет) и неизбежно ведет к смерти на фоне нарастающих психических и двигательных расстройств. В крови и спинномозговой жидкости определяются высокие титры противокоревых антител. В дегенерирующих нейронах можно найти коревые белки и РНК, а сам вирус удается выявить при сокультивировании с чувствительными клетками. По ряду признаков ПСПЭ-вирус отличается от классического вируса кори, являясь его дефектным вариантом с ослабленным репликативным потенциалом. Это связано с рядом мутаций, главная из которых вызывает дефект по М-белку, препятствующий созреванию вирионов.
Вирус кори способен преодолевать плацентарный барьер, но данные о последствиях внутриутробной инфекции противоречивы. Пороки развития и гибель плода, если и возникают, то лишь в первом триместре беременности. Тем не менее серонегативные (лишенные противокоревых антител) беременные женщины, оказавшиеся в очаге коревой инфекции, должны быть под специальным наблюдением.
В типичных случаях корь продолжается 7—11 дней, из которых 2—4 дня приходится на продромальный (катаральный) период и 5—7 дней — на экзантемную фазу. Хотя выделение вируса начинается в конце инкубационного периода и продолжается до 5 дней (чаще 2—3 дня) после появления сыпи, наибольшую эпидемическую опасность представляют больные в продромальном периоде (катаральная фаза). Именно в это время назофарингеальный секрет содержит максимальное количество вируса, а кашель усиливает вероятность его распространения. Поэтому распространение инфекции часто происходит до постановки диагноза. Кроме назофарингеального секрета вирус содержится в слезах, крови и моче, отражая не только гематогенную диссеминацию, но и политропную репликацию вируса.
Иммунитет. На модели кори хорошо прослеживается значение различных иммунных механизмов в предупреждении, выздоровлении и патогенезе вирусных инфекций. Перенесенная корь оставляет пожизненный иммунитет к реинфекции. Его основой являются антигенная монолитность и консерватизм вируса, который, как уже говорилось, представлен единственным серотипом. Корь сопровождается образованием и длительным сохранением IgG-антител и различных вариантов сенсибилизированных Т-лимфоцитов. В респираторных секретах обнаруживаются IgA-антитела.
Представления о протективной функции антител зародились в начале 1920-х гг. Это нашло отражение в специфической профилактике кори путем введения сыворотки здоровых людей, в которой обычно содержатся противокоревые антитела — постинфекционные или поствакцинальные. Позже вместо цельной сыворотки стали использовать гамма-глобулиновую фракцию — нормальный иммуноглобулин человека, долго именовавшийся противокоревым гамма-глобулином. Его широкое применение в допрививочный период утвердило за антителами репутацию надежного эффектора противокоревого иммунитета. Вместе с тем было замечено, что профилактическое действие иммуноглобулина проявляется только в первые 5—7 дней инкубационного периода; как лечебный препарат он практически бесполезен.
Неожиданными оказались и наблюдения за детьми с врожденной агаммаглобулинемией, не способными продуцировать антитела. По чувствительности к кори, ее течению и исходам они не отличаются от здоровых детей. Более того, такие дети обретают надежный иммунитет к реинфекции. Вместе с тем, дефекты клеточного иммунитета делают заболевание исключительно опасным, предрасполагая к гибели от коревых осложнений, чаще от гигантоклеточной пневмонии. Это говорит о том, что антитела — не главный и не единственный фактор, определяющий взаимоотношения с вирусом кори. Внося существенный вклад в защиту от инфекции на этапе первичной вирусемии, они бессильны против реплицирующегося вируса, который циркулирует в составе лейкоцитов, распространяясь при помощи тесных межклеточных контактов (симпласты!).
По современным представлениям, основная роль в «поздней» профилактике и выздоровлении принадлежит эффекторам клеточного иммунитета — CD8+ и CD4+ (Th1)Т-лимфоцитам. Первые напрямую убивают клетки, экспрессирующие вирусные антигены, вторые достигают этого опосредованно, содействуя дифференцировке цитотоксических CD8+-лимфоцитов и привлекая эффекторы иммунного воспаления. В условиях перегрузки вирусными антигенами такие реакции выходят из-под гомеостатического контроля, проявляясь в виде клинически значимого синдрома. Особенно ярко это выражено при коревой сыпи. Ее центральным элементом служит очаг иммунологически зависимого воспаления вокруг сосуда, эндотелиоциты которого инфицированы вирусом, являясь мишенью для Т-лимфоцитов. Они запускают каскад реакций, которые обостряют и усиливают альтерацию сосудистой стенки, вызывая кровоизлияния в кожу (экзантема) или слизистую оболочку (энантема). На этой основе формируется механизм саногенеза, который, убивая инфицированные клетки, уничтожает и вирус. В иммунном организме, перенесшем естественную или вакцинальную (живая вакцина) инфекцию, Т-зависимая элиминация зараженных клеток опережает репликацию вируса, обрывая накопление его патогенетически значимой массы. В отличие от антител, протективность которых базируется на взаимодействии с поверхностными антигенами вириона, в антивирусных реакциях Т-лимфоцитов задействованы и внутренние антигены, экспрессирующие свои эпитопы на поверхности зараженных клеток в составе молекул главного комплекса гистосовместимости.
Взаимоотношения вируса кори с иммунной системой далеки от ортодоксальности. Коревая инфекция сопровождается бепрецедентной блокадой реактивности к антигенам. Символом этого является подавление реакций гиперчувствительности замедленного типа, в частности аллергии к туберкулину. Ослабление иммунитета предрасполагает к суперинфекциям и активации латентных инфекций. Не случайно при кори наблюдается обострение туберкулеза и герпетических инфекций. Очевидно, по той же причине корь снижает агрессивность иммунологически зависимых заболеваний, в частности, бронхиальной астмы. Но все это носит временный характер, и примерно через месяц иммунный статус восстанавливается. Причины коревой иммунодепрессии требуют уточнения, но ясно, что они связаны не с повреждением, а с функциональными перекосами в системе Т-клеточного иммунитета. Согласно концепции о поляризации CD4+ Т-хелперов (Тh) коревая инфекция побуждает к усиленной секреции цитокинов Тh2-типа, которые тормозят Th1-зависимую дифференцировку Т-эффекторов, ослабляя реакции клеточного иммунитета. Из этой логики выпадает, однако, сам возбудитель: не понятно, почему блокада распространяется только на чужие антигены, реактивность к вирусу кори сохраняется.
Вакцинация. В первой коревой вакцине был использован вирус, убитый формалином. Ее эффект был не только неубедительным (скорее всего из-за отсутствия образования анти-F-антител), но и неожиданным: на поствакцинальном фоне инфекции протекали тяжелее, чем у невакцинированных, напоминая историю с вакцинацией против респираторно-синцитиального вируса (см. выше). Первая живая вакцина (1963 г.) была недостаточно аттенуирована и часто вызывала настоящую корь, поэтому ее приходилось вводить в комплексе с иммуноглобулином (еще одно доказательство протективности антител!). Современные вакцинные штаммы аттенуированы лучше, реже дают побочные реакции и обладают высокой превентивностью. Иммунизируют годовалых детей, обычно в комбинации с вакцинами против краснухи и паротита. Ревакцинация предусмотрена в 6 лет, но она показана только серонегативным детям (на практике такой отбор организовать трудно).
Классическую корь в довакцинальном периоде определяли эпидемии продолжительностью 2—3 мес, которые происходили в зимне-весенний период в популяциях больших городов с интервалом 2—4 года. Около 90% случаев падало на возраст до 10 лет, после 30 лет болезнь практически не встречалась. Вакцинация изменила картину, увеличив интервалы между эпидемиями (или заменив их спорадической заболеваемостью), сократив продолжительность вспышек и сместив акцент на более старший возраст. Увеличился риск заболеваемости среди детей первого года жизни, что объясняется недостатком материнских антител на фоне резкого снижения циркуляции вируса, которая необходима для подстегивания поствакцинального иммунитета.
Полагают, что для создания зон, свободных от болезни, требуется вакцинация более 90% детей. По мере приближения к 100% вирус, лишенный условий для репликации, должен быть вытеснен из популяции. Это определяет стратегию глобального искоренения вируса в условиях поголовной вакцинации детского населения Земного шара. Однако, кроме технических (точнее экономических) проблем, есть принципиальные соображения, вносящие диcсонанс в эту концепцию. Речь идет о возможности резервации вируса кори на фоне напряженного коллективного иммунитета. Признание этого означает, что для самоподдержания коревому вирусу не требуется клинически значимого конфликта с хозяином, а достаточно субклинических контактов, которые возникают на фоне протективного иммунитета, сдерживающего инвазию вируса, но позволяющего ему некоторое время бессимптомно реплицироваться в зоне входных ворот инфекции. Этому, казалось бы, противоречит то, что кроме антиинвазивного иммунитета коревая инфекция и вакцинация оставляют местный иммунитет, действующий на уровне слизистых оболочек верхних дыхательных путей. Но это главным образом IgA-антитела, которые имеют короткую память, увядая гораздо быстрее, чем IgG-зависимый и клеточный иммунитеты. Данное обстоятельство может привести к разобщению устойчивости к инвазии (вирусемии) и местной колонизации.
Без генерализации коревая инфекция бессимптомна, но она может быть достаточной для выживания вируса. По крайней мере случаи инаппарантной инфекции вакцинированных неоднократно наблюдались во время коревых вспышек. Вопрос в том, сможет ли вирус длительно просуществовать при столь ограниченной активности или будет полностью вытеснен, как это случилось с вирусом натуральной оспы. Для большинства регионов корь остается эндемичной инфекцией, хотя в ряде стран (США, Финляндия, некоторые страны Центральной и Южной Америки) эндемичная передача вируса, как будто, прекращена. Вместе с тем рецидивы кори даже в странах с хорошо налаженными противокоревыми прививками говорят о том, что полной и длительной стабилизации коллективного иммунитета не произошло. Есть надежда, что этого удастся достичь при более интенсивной вакцинации. В последние годы оформилось понятие «возрождающиеся инфекции» (англ. reemerging infections). Об этом следует помнить, приветствуя победы над опасными болезнями. Корь может быть одной из них.
Лабораторная диагностика. При цитологическом исследовании респираторного, конъюнктивального секретов или осадка мочи можно обнаружить многоядерные гигантские клетки с характерными внутриядерными включениями. При помощи иммунофлюоресценции в них выявляются коревые антигены. Для серологического подтверждения требуется четырехкратное нарастание титров антител в динамике заболевания (между острой фазой и фазой выздоровления, т.е. в первые 3—4 дня после начала сыпи и 3—4 нед спустя). Лучше всего подходит реакция торможения гемагглютинации, хотя можно воспользоваться связыванием комплемента и нейтрализацией вируса. При однократном анализе требуется обнаружение IgM-антител, которые выявляются у 70% больных к началу высыпаний и почти у 100% на 3—4-й дни сыпи. Разработаны способы выявления вирусных генов, в том числе непосредственно в зараженных клетках. Впрочем, при классическом течении кори диагноз не требует лабораторного подтверждения; он полезен лишь в атипичных случаях.