
2006 год
А.Н. Маянский. Патогенетическая микробиология: руководство. Нижний Новгород: Издательство НижГМА, 2006. – 520 с., ил. ISBN 5-7032-0643-Х. Рекомендуется Учебно-методическим объединением по медицинскому и фармацевтическому образованию вузов России в качестве учебного пособия для системы послевузовского профессионального образования врачей.
Книга посвящена актуальным проблемам инфекционной патологии. На основе взаимоотношений в системе «хозяин-паразит» анализируются свойства бактерий, грибов, вирусов, которые вносят существенный вклад в развитие микробных болезней. По сравнению с предыдущим изданием (Микробиология для врачей. Н. Новгород: Издательство НГМА, 1999) существенно обновлен раздел о болезнетворности микроорганизмов. С учетом новых научных данных внесены исправления в главы о воспалительном процессе, диагностике инфекционных заболеваний, апоптозе, дисбиотических состояниях и нормальной микрофлоре. Новые очерки посвящены микобактериям туберкулеза, шигеллам, сальмонеллам, холерному вибриону, общей микологии, парамиксовирусам, рино- и коронавирусам, аденовирусам, папилломавирусам, вирусу бешенства, арбо- и родентвирусам. Основное внимание сконцентрировано на патогенетических принципах микробиологии, которые сближают научные факты с практической медициной, позволяя ориентироваться в сложных взаимодействиях микробов и человека.
Для врачей, преподавателей и студентов медицинских вузов.
Предисловие
Патогенность микроорганизмов — частный случай их экологии.
Микробиология — наука о живых объектах, неразличимых невооруженным глазом. Человек не воспринимает частицы размером менее 0,1 мм, и именно это служит критерием для разделения организмов на микро- и макробиологические, или просто биологические.
Все, что меньше 0,1 мм, принадлежит микробиологии; все, что больше — биологии. Условность такой категоричности очевидна. Во-первых, микробы по базисной физиологии похожи на высшие организмы, что отражает единство живой материи.
А во-вторых, подобная трактовка делает мир микробов необозримо многоликим. Своеобразие его отдельных представителей значительно превосходит различия между биологическими объектами. Достаточно вспомнить об уникальности генетического материала РНК-содержащих вирусов!
Экологическая пластичность гарантирует микробам убиквитарность. Это служит сохранению жизни на нашей планете, обеспечивая непрерывность цепей питания, сопрягающих все живое.
Ключевым моментом является минерализация органогенов, которые в виде мертвых остатков поступают в природу после отмирания растений, животных и самих микробов. Растения (почти единственные созидатели органической материи) являются автотрофами и используют неорганические источники органогенов. Животные прямо или косвенно потребляют соединения, синтезированные растениями. Умирая, и те и другие оставляют материал, который напрямую не может быть возвращен в круговорот биосферы.
Земля не покрывается трупами растений и животных только потому, что существует еще одна, формально невидимая жизнь — жизнь микробов. Бактерии, производящие брожение и гниение, переводят углерод, азот, водород, кислород и фосфор в форму, пригодную для растений, т.е. для созидания нового живого материала. Перефразируя А.М. Горького, можно сказать, что благодаря микроорганизмам смерть покорно служит делу жизни.
В медицинской микробиологии представлена лишь малая часть царства микробов — те из них, которые вступают в непосредственные взаимоотношения с человеком. Противоречие между хозяином и паразитом рождает инфекционный процесс, который находит клиническое выражение либо ограничивается внутренним конфликтом, о котором судят по иммунным сдвигам в зараженном организме.
Поэтому медицинская микробиология включает два раздела — исследование инфекционных агентов и изучение реактивности организма, основанной на врожденном и приобретенном иммунитете. В своей книге мы стремились следовать этой идеологии.
Представленный материал является обновленным и существенно расширенным изданием книги «Микробиология для врачей», опубликованной издательством НГМА в 1999 г. Эта книга быстро разошлась среди читателей, получив добрые официальные и неофициальные отзывы.
Название новой книги («Патогенетическая микробиология») отражает авторский выбор излагаемых сюжетов, а также сущность предмета, отношение к взаимодействию микробов с человеком, который не только создает жизненную среду для инфекционных агентов, но и сам испытывает на себе их действие. Немало пришлось изменить, учитывая современные достижения науки, но многое удалось и сохранить. Написан ряд новых очерков, посвященных микобактериям туберкулеза, шигеллам, сальмонеллам, холерному вибриону, микологии, парамиксовирусам, рино- и коронавирусам, аденовирусам, папилломавирусам, вирусу бешенства, арбо- и родентвирусам.
Автор с благодарностью воспримет все конструктивные замечания и пожелания в надежде, что они послужат развитию концептуальных основ микробиологии.
Бог изощрен, но не злонамерен.
А. Эйнштейн
Болезнетворность бактерий
- Категории болезнетворности.
- Адгезия и колонизация.
- Инвазия.
- Бактериальные токсины.
- Прямая и опосредованная токсичность.
- Персистенция.
- Микробные сообщества (биопленки).
- Генетика болезнетворности.
В зависимости от взаимоотношений с хозяином микробы делятся на непатогенные, патогенные и условно-патогенные. Если безвредность первых практически абсолютна, а патогенные виды облигатно болезнетворны, то реализация условной патогенности решается в индивидуальных системах «макро—микроорганизм», так как зависит от резистентности хозяина.
Повышенная чувствительность характерна для лиц с ослаблением иммунитета, — местного или общего, специфического или неспецифического. Таких больных принято называть иммунокомпрометированными, а возбудители, которым они не оказывают должного сопротивления, — микробами-оппортунистами, или оппортунистическими патогенами (от англ. opportunity — удобный, подходящий случай). Нередко иммунитет компрометируют врачебные процедуры, например использование иммунодепрессантов или инструментальных вмешательств, нарушающих целостность внешних покровов. Поэтому большинство оппортунистических инфекций относится к внутрибольничным (госпитальным) осложнениям, определяя главное содержание клинической микробиологии.
Едва ли не все из микробов-оппортунистов постоянно или временно входят в состав нормальной микрофлоры, создавая прецедент для эндогенных, или аутоинфекций.
Патогенные микроорганизмы опасны для практически здоровых людей. К ним относятся возбудители классических инфекционных заболеваний, с которыми имеют дело врачи специализированных (инфекционных) стационаров. Они всегда возникают при экзогенном заражении и не привязаны к госпитальным условиям, так как иммуносупрессия не обязательна для их развития. Способность поражать здоровых является причиной групповой заболеваемости, вплоть до эпидемий.
Сочетание высокой контагиозности с тяжелым (опасным для жизни) течением определяет принадлежность микроорганизмов к возбудителям особо опасных инфекций.
Понятия «условная» и «безусловная» патогенность во многом утилитарны, и грань между ними не всегда убедительна. Болезнетворность даже самых опасных микробов зависит от определенных обстоятельств: дозы инфекта, пути инфицирования, выживаемости во внешней среде, наконец, от восприимчивости организма, лишенного видового иммунитета.
Непросто, например, решить вопрос о принадлежности микроорганизма к одной из этих категорий, если для возбуждения патологического процесса требуется проникновение бактерий через микротравмы кожи и слизистых оболочек. Формально здесь налицо нарушение иммунитетного барьера, но фактически микроб сталкивается со здоровым организмом.
Впрочем, любая классификация допускает исключения. Важен принцип, и здесь он базируется на признании того, что патогенность — потенциальный признак, который раскрывается при определенном стечении обстоятельств. Их должно быть много для одних микробов и меньше для других. Этим определяется позиция на шкале болезнетворности — от безвредных комменсалов до возбудителей особо опасных инфекций.
Патогенность отражает болезнетворность на уровне вида. Признак подвержен внутривидовым колебаниям, т.е. неодинаково выражен у разных штаммов (в генетической интерпретации — клонов) бактерий. В связи с этим используется еще одно понятие — вирулентность, которое отражает степень болезнетворности штаммов внутри патогенного вида. Путем генетических преобразований даже для возбудителей особо опасных инфекций могут быть получены авирулентные варианты, и, наоборот, безвредные бактерии можно сделать носителями генов вирулентности.
Болезнетворность — многофакторный признак. Это логично, если учесть многоликость защитных механизмов, которые обязан преодолеть инфекционный агент, претендующий на агрессию. Из базисных факторов, определяющих болезнетворность, выделим следующие:
- способность к адгезии/колонизации зоны первичного инфицирования (входных ворот инфекции);
- способность к инвазии, т.е. к выходу за пределы зоны первичной колонизации;
- устойчивость во внешней среде и способность к распространению;
- токсигенность;
- способность к персистенции.
Адгезия/колонизация
Колонизация означает закрепление и последующее размножение бактерий в зоне инфицирования. Для инфекций, которые начинаются со слизистых оболочек (их большинство), первым и необходимым этапом является преодоление колонизационной резистентности мукозального тракта.
Процесс начинается с адгезии, основанной на избирательном взаимодействии бактерий с рецепторами эпителиоцитов и/или покрывающим их слоем слизи (муцинами). Некоторые бактерии активно готовят участки для закрепления, обнажая клеточные рецепторы (например, при помощи нейраминидазы), пользуются вирусиндуцированной и/или цитокининдуцированной экспрессией эпителиальных рецепторов, дефектами в эпителиальном покрове, сорбируются на других бактериях, ранее обосновавшихся в данном биотопе, или взаимодействуют с растворимыми белками (например, фибронектином, витронектином), опосредуя через них закрепление на клеточных рецепторах и в соединительнотканном матриксе.
Спектр бактериальных адгезинов обширен, что определяет поливалентность феномена даже в масштабе одного вида. У грамотрицательных бактерий в нем участвуют микроворсинки (пили), белки и липополисахариды наружной мембраны, у грамположительных — белки клеточной стенки, липотейхоевые кислоты; у капсульных бактерий в адгезию включаются капсульные полисахариды. Большинство наблюдений сделано в клеточных культурах и весьма условно отражают поведение бактерий в реальных ситуациях. Однако сам факт патогенетически значимой адгезии не вызывает сомнений. В таких случаях она выходит за рамки чисто физического феномена, являясь одним из факторов экологически значимой детерминации вирулентности (см. ниже).
Уместно говорить о контактной болезнетворности, включая сюда изменения клеток, инициируемые микробной адгезией. В ее реализации участвуют два основных механизма:
1) медиаторные сигналы, запускаемые через клеточные рецепторы, ангажируемые бактериальными лигандами (адгезинами);
2) действие бактериальных белков, инъецируемых внутрь клетки (отсюда — инъекционные токсины).
В обоих случаях происходит перестройка внутриклеточного гомеостаза (через активацию сигнальных каналов клетки-хозяина), выгодная возбудителю.
Широко распространены извращения цитоскелета, ведущие к индуцированному фагоцитозу и макропиноцитозу. Это обеспечивает проникновение бактерий в нефагоцитирующие клетки, в частности эпителиоциты и эндотелиоциты. За поглощением может последовать трансцитоз (т.е. прохождение эпителиального барьера) и распространение возбудителя на соседние клетки. Так ведут себя шигеллы, сальмонеллы, йерсинии, менингококки, гонококки и др.
Поразительной изобретательностью обладают шигеллы, листерии и риккетсии. Оказавшись внутри клеток, они используют актин для построения псевдожгутика, получая возможность для активного передвижения внутри и между клеток.
Функциональная перестройка клеток, контактирующих с бактериями и их продуктами, может сопровождаться секрецией провоспалительных цитокинов и других флогогенных медиаторов. Это предрасполагает к развитию пиогенных воспалительных реакций (без классического бактериального повреждения). Такой механизм входит в патогенетический потенциал ряда бактерий, инфицирующих слизистые оболочки (уропатогенные эшерихии, гонококк и др.).
Адгезия создает предпосылки для размножения бактерий. Чтобы добиться этого, они должны выстоять против биоцидных и биостатических факторов, которые в разных количествах и сочетаниях представлены в секретах мукозального тракта. Среди паразитов слизистых оболочек распространена способность к продукции IgA-протеаз, которые расщепляют IgA-антитела, лишая их антиадгезивного эффекта. Многие бактерии нейтрализуют лизоцим, инактивируют оксиданты и другие потенциально вредные начала, продуцируют сидерофоры, конкурируя с лактоферрином за ионы железа. Удивительной стойкостью, например, обладает Helicobacter pylori: он активно персистирует в желудке, нейтрализуя вокруг себя кислую среду благодаря высокой уреазной активности.
Для питания хозяином предоставляются широкие возможности, хотя за реализацию некоторых из них бактериям приходится бороться. Один из базисных механизмов — конкуренция за железо. Его отсутствие гибельно для бактерий, поскольку блокируется построение окислительно-восстановительных ферментов.
Ионы железа прочно связаны с белками хозяина, например с ферритином, трансферрином, лактоферрином и гемоглобином. Бактерии пользуются разными (часто несколькими) способами, чтобы обеспечить себя этим важнейшим фактором роста. Они отнимают железо у железосвязывающих белков, «вылавливая» их при помощи поверхностных рецепторов (например, трансферриновые и лактоферриновые рецепторы менингококка, гонококка и палочки инфлюэнцы, связывание гемоглобина холерным вибрионом); продуцируют белки (сидерофоры), конкурирующие за свободное железо с белками хозяина (вибриобактин холерного вибриона, пиовердин и пиохетин синегнойной палочки, сидерофоры кишечной палочки, шигелл, сальмонелл); высвобождают связанное железо путем протеолиза с его последующей фиксацией сидерофорами (синегнойная палочка).
Инвазия
Внутриэпителиальная инвазия. Колонизация слизистых оболочек не всегда ограничивается их поверхностью. Патогенность ряда бактерий связана с проникновением (пенетрацией) в эпителиальные клетки, где они размножаются, вызывая повреждение (шигеллы, йерсинии, гонококк, хламидии и др.).
Способность к пенетрации определяется особыми факторами, среди которых лучше всего охарактеризованы так называемые инвазины. Это адгезины бактерий, которые, взаимодействуя с рецепторами клеток (интегринами), запускают внутриклеточные сигналы, индуцирующие поглощение бактерий. По сути, это вариант контактной болезнетворности, о которой говорилось выше. На модели йерсиний (Y. pseudotuberculоsis) удалось идентифицировать ген инвазина, передав этот признак непенетрирующему штамму кишечной палочки.
Субэпителиальная инвазия. Нередко болезнетворность проявляется после размножения бактерий в субэпителиальных тканях. Фактически это та же колонизация, но уже иных тканей и в иных условиях.
Разные бактерии неодинаково решают проблему прохождения через внешние покровы. Многие из них, особенно бактерии-оппортунисты, неспособны к обильной колонизации и активной пенетрации слизистых оболочек и кожи. Для этого они пользуются пассивной инвазией, проникая через повреждения эпителия, например после травматичных врачебных процедур (абиогенная трансмиссия).
Пассивная инвазия — практически единственный способ преодоления кожного барьера, так как неповрежденный эпидермис непроницаем для большинства бактерий. Кроме микротравм и инородных тел микробы научились использовать кровососущих насекомых — биогенная трансмиссия. Вариантом пассивной (точнее, спровоцированной) инвазии являются вторичные инфекции, которые развиваются на фоне вирусных поражений эпителия (например, бактериальные пневмонии при гриппе) или осложняют другие (первичные) бактериальные инфекции (например, коклюш, гонорею).
Активная инвазия характерна для возбудителей ряда мукозальных инфекций. С этой целью некоторые из них используют особые М-клетки, покрывающие лимфоидные фолликулы. Они были впервые обнаружены над пейеровыми бляшками и получили свое название из-за особенностей мембраны (отсутствие щеточной каймы и тонкий слой гликокаликса). Избирательность их локализации объясняется тем, что дифференцировка М-клеток из стволовых клеток эпителия возбуждается цитокинами лимфоидных фолликулов. Лишенные лизосомальных протеаз, М-клетки являются удачным объектом для трансцитоза нативных макромолекул и бактерий в субэпителиальную ткань. Кроме тонкого и толстого кишечника М-клетки обнаружены в эпителиальном покрове миндалин, аденоидов и респираторного тракта.
Внутриэпителиальная инвазия и трансцитоз являются импульсом, побуждающим эпителиальные клетки к секреции цитокинов (хемокинов), вызывающих воспаление — начало патологического процесса.
Оказавшись во внутренней среде (в нашем контексте — под эпителиальным покровом), возбудители сталкиваются с еще более сложным набором факторов и механизмов иммунитета.
При стабилизации и начавшемся размножении бактерий главным антиинвазивным механизмом служит воспалительная реакция, которая обеспечивает мобилизацию и взаимодействие многих иммунитетных ресурсов. Центральным эффектором, нацеленным на уничтожение «вредных деятелей», являются фагоциты, действующие в содружестве с гуморальными факторами — хемоаттрактантами и опсонинами (см. «Механизмы противоинфекционного иммунитета»).
Отсюда первоочередная задача бактерий — задержать или ослабить фагоцитарную реакцию. Неслучайно врожденные дефекты нейтрофилов и ассистирующих им факторов (комплемента, иммуноглобулинов) чреваты серьезными осложнениями.
Бактерии оказывают сопротивление на всех этапах фагоцитоза — от хемотаксиса до внутрифагосомального уничтожения. Для этого они располагают богатым тактическим арсеналом, который включает продукты, действующие на клетки и гуморальные факторы. Начать с того, что бактерии разрушают хемоаттрактанты (например, С5а) и продуцируют токсины, убивающие фагоциты (например, лейкоцидины золотистого стафилококка, фосфолипазы клостридий). Взаимодействие с фагоцитами блокируется капсулой и другими высокогидрофильными поверхностными структурами бактерий. Некоторые бактерии сорбируют белки хозяина, обретая нечто вроде псевдокапсулы (например, фибриновая пленка коагулазопозитивных стафилококков). Известны проявления антиопсонической активности в системе «нейтрофилы—антитела—комплемент». Некоторые из бактериальных продуктов (например, аденилатциклазы) действуют как фармакологические агенты: они не повреждают фагоциты, но извращают их функции, ослабляя хемотаксис, поглощение и образование фаголизосом.
Новые представления о патогенезе инфекционных заболеваний связаны с концепцией апоптоза. Они затрагивают и механизмы антифагоцитарной активности бактерий, среди которых есть такие, которые способствуют преодолению антимикробного арсенала фагоцитов. Это ослабляет эффективность фагоцитарной атаки, повышая вероятность выживания патогенов.
Но даже внутри фаголизосом бактерии не безоружны: они инактивируют биоагрессивные метаболиты кислорода (супероксиддисмутаза), подавляют действие пероксидазы (каталаза), экранируют собственную ДНК от оксидантов (антиоксидантные белки) и др.
Все это препятствует фагоцитозу или делает его незавершенным. Буксуя, фагоциты выделяют комплекс деструктивных элементов, вынужденно включаясь в аутоагрессию, т.е. в повреждение собственных тканей хозяина. В расширении зоны инвазии участвуют и бактериальные ферменты, которые разрушают основное вещество соединительной ткани и базальную мембрану эндотелия (гиалуронидаза, эластаза, коллагеназа), устраняют механический барьер из сгустков фибрина (фибринолизин), разжижают гнойный экссудат (ДНК-аза, протеиназы).
Внутрисосудистая инвазия (бактериемия). Попав в кровяное русло, бактерии быстро погибают или фагоцитируются резидентными макрофагами. Но есть и такие, которые в этих условиях выживают. Более того, в ряде случаев бактериемия является обязательной фазой инфекционного процесса (возбудители брюшного тифа, менингита, бруцеллеза и др.).
Чтобы выстоять, бактерии должны прежде всего «уйти» от комплемента, биоцидность которого многократно усиливается антителами. Это особенно важно для грамотрицательных форм, высокочувствительных к литическому действию мембраноатакующего комплекса комплемента. Без поддержки антител комплемент почти бессилен против инкапсулированных бактерий (менингококк, пневмококк, палочка инфлюэнцы, тип b), тем более, что благодаря структурным особенностям капсульных полисахаридов некоторые из них избегают активации комплемента по альтернативному (антителонезависимому) пути. Сальмонеллы и бруцеллы вносятся в кровь инфицированными лимфоцитами, которые защищают их от контакта с бактерицидными факторами крови. Уверенно чувствует себя золотистый стафилококк: он циркулирует внутри микротромбов, образующихся под влиянием его коагулазы.
В целом, выживание в крови создает дополнительные шансы для реализации патогенетического потенциала бактерий: находя подходящие условия внутри сосудистого русла (повреждение эндотелия, тромбы, внутрисосудистые протезы) или в тканях, они формируют вторичные очаги инфекции, которые эволюционируют согласно общим принципам колонизации/инвазии.
Внутримакрофагальная инфекция. Одним из надежных способов избежать контакта с гуморальными эффекторами иммунитета (антителами и комплементом) является внутриклеточное паразитирование. По ряду причин (см. «Механизмы противоинфекционного иммунитета») стратегически оптимальным является паразитирование внутри макрофагов.
Мононуклеарные фагоциты используются рядом бактерий, грибов и простейших как экологическая ниша, из которой они угрожают хозяину. Для этого возбудитель должен решить непростую задачу — подавить потенциальную агрессивность фагоцитов или устоять против их биоцидных факторов. Внутримакрофагальные паразиты добиваются этого разными способами. Оказавшись внутри фагосом, они блокируют их слияние с лизосомами, где сосредоточен биоцидный резерв фагоцитов (микобактерии туберкулеза, токсоплазмы, гистоплазмы, легионеллы), лизируют фагосомы фосфолипазными ферментами, ускользая в цитоплазму (микобактерии туберкулеза и лепры, листерии, риккетсии, трипаносомы), ослабляют закисление внутри фагосом (сальмонеллы).
Вместо активации, которая по логике должна сопутствовать фагоцитозу, макрофаги в таких случаях не реагируют на присутствие паразита, нуждаясь в дополнительных стимулах, чтобы выйти из эффекторного паралича (см. «Механизмы противоинфекционного иммунитета»).
Кроме того, некоторые микробы (например, микобактерии туберкулеза) устойчивы к биоцидным факторам фагоцитов, сохраняя жизнеспособность даже в активированных макрофагах, которые не только не обеспечивают эффективной элиминации возбудителя, но и содействуют деструкции тканей. Определенное значение имеет и индукция апоптоза зараженных макрофагов, который выключает их из активной антимикробной деятельности.
Интоксикация
Токсины (греч. toxicon — яд) — продукты жизнедеятельности бактерий, которые в малых дозах вызывают структурные или функциональные повреждения клеток. Интоксикация является главным механизмом инфекционного процесса, а токсины — ведущим фактором болезнетворности бактерий. Они оказывают не только местное воздействие, способствуя закреплению инфекта и повышая вероятность инвазии, но и вызывают системные эффекты, далеко выходящие за зону первичной колонизации.
История изучения бактериальных токсинов началась в 1888 г., когда Э.Ру и А.Иерсен (сотрудники Л.Пастера) отделили токсическое начало дифтерийной палочки от бактерий и воспроизвели основные симптомы дифтерийной инфекции на животных при помощи бесклеточного фильтрата. Это инициировало множество исследований с другими бактериями, ознаменовав рождение новой отрасли микробиологии — токсинологии. Успех Э.Ру и А.Иерсена был повторен на модели возбудителей столбняка, ботулизма, холеры и ряда других инфекций. Но, как обычно, реальность оказалась сложнее.
Далеко не всегда патогенез инфекционного процесса укладывается в рамки мономолекулярной интоксикации, хотя токсический компонент сопутствует практически всем инфекционным заболеваниям.
Характеристика токсинов
Токсины бактерий принято делить на две категории: экзо- и эндотоксины. Экзотоксины чаще обнаруживаются в фазе активного роста и аккумулируются в цитоплазме, выделяясь путем активной секреции либо при аутолизе бактериальных клеток (например, у клостридий). Эндотоксины входят в состав клеточной стенки, высвобождаются после разрушения бактерий и накапливаются в среде при отмирании клеток.
Экзотоксины относятся к белкам и обычно секретируются в виде предшественников — протоксинов. Их активация связана с ограниченным протеолизом эндопептидазами клеток-мишеней и/или собственными ферментами бактерий. Классические эндотоксины являются липополисахаридами (ЛПС) наружной мембраны грамотрицательных бактерий, построенными по общей схеме. Полисахаридный фрагмент представлен кор-компонентом и О-радикалом, который теряется у R-вариантов. С полисахаридом связаны антигенные свойства молекулы, причем О-радикал обладает эпитопным полиморфизмом даже внутри одного вида бактерий (О-серовары). Кор-компонент относительно консервативен: количество его сероваров ограничено даже в масштабе крупных таксонов, определяя перекрестные серологические реакции (например, у энтеробактерий). Липид А отвечает за токсичность молекулы.
Грамположительные бактерии лишены наружной мембраны, а вместе с ней и липополисахаридного эндотоксина. Однако некоторые компоненты их клеточной стенки (пептидогликан, тейхоевые кислоты) обладают биологическими свойствами ЛПС, хотя в равных концентрациях менее активны.
Информация об эндотоксинах, как и о всех структурных компонентах клетки, заложена в «собственных» (хромосомных) генах бактерий. Напротив, способность к образованию экзотоксинов часто детерминируют внехромосомные гены — tox-гены плазмид и умеренных фагов. По этому поводу У.Хэйс, автор классической монографии по генетике бактерий и бактериофагов, остроумно заметил: «Остается только гадать, сколько раз мы несправедливо взваливали на бактерии грехи их вирусов».
Биологический смысл токсинообразования неясен. Во всяком случае, можно получить бактерии-двойники, лишенные способности к продукции токсинов, но не страдающие от этого. Вместе с тем, эволюционная стабильность токсинообразования побуждает думать, что значение этого признака не исчерпывается агрессией против хозяина.
Активность бактериальных токсинов колеблется в широких пределах. Особенно действенны экзотоксины. Для самого мощного из них, ботулинического, хватило бы 6 кг, чтобы истребить все человечество. Миллиграмм столбнячного токсина способен убить 150 млн. мышей, а такое же количество токсина дифтерийной палочки смертельно для 150 тыс. морских свинок.
Токсичность ЛПС и эндотоксиноподобных продуктов грамположительных бактерий намного ниже и проявляется в условиях, обеспечивающих всасывание значительной порции токсических веществ. Впрочем, есть примеры катастрофического повышения чувствительности к ЛПС. Это моделируется в знаменитом феномене Шварцмана — диссеминированной, обычно смертельной коагулопатии после повторного (через 24 ч) введения ЛПС. Вместо ЛПС для разрешающей инъекции могут быть использованы другие продукты бактериальной и небактериальной природы, что отражает неспецифическую природу реакции и широкий спектр сенсибилизирующей активности эндотоксинов. Добавим, что ЛПС усиливают токсичность скарлатинозного токсина и, возможно, других суперантигенов (см. ниже).
В проекции на инфекционный процесс можно говорить о токсинах, которые определяют клиническую картину заболевания, и о токсинах, не имеющих четкой патогенетической доминанты. К первым из них относятся токсины дифтерийной палочки, холерного вибриона, клостридий, вызывающих столбняк и ботулизм, возбудителя сибирской язвы, энтеротоксигенных энтеробактерий, некоторых стафилококков и стрептококков. Токсинов второго типа гораздо больше. Их называют «парциальными токсинами» или «токсинами частного приложения», подразумевая, что, действуя раздельно, они не определяют специфики патологического процесса.
Кроме того, в интоксикации принимают участие вторичные токсины, которые выделяются при повреждении или активации клеток хозяина, что особенно характерно для ЛПС, вызывающих гиперсекрецию флогогенных цитокинов из стимулированных макрофагов и других клеток. Наведенным токсическим эффектом обладают суперантигены, неспецифически стимулирующие Т-лимфоциты (см. ниже).
Названия экзотоксинов традиционно базируются на категории поражаемых мишеней, хотя это не всегда совпадает с масштабом патогенетического эффекта. Ниже приведена терминология, которая с известной долей условности отражает биологическую активность бактериальных токсинов.
Энтеротоксины — поражают эпителий (энтероциты) тонкого кишечника; иногда под эту рубрику попадают токсины, проникающие через кишечник, но не оказывающие на него прямого действия (например, энтеротоксины золотистого стафилококка).
Нейротоксины — вызывают избирательное поражение нервных клеток, межнейронных и нервно-мышечных синапсов.
Дермонекротоксины — поражают кожу. К этой весьма расплывчатой группе относятся эксфолиатины золотистого стафилококка, обладающие уникальной способностью разрушать эпителиальные десмосомы (см. «Стафилококк»).
Гемолизины — лизируют эритроциты. Определение нельзя понимать буквально, так как цитотоксичность гемолизинов не ограничивается лизисом эритроцитов, распространяясь на другие типы клеток. Эритроциты — лишь удобная модель для изучения цитолитического эффекта.
Лейкоцидины — антифагоцитарные факторы, повреждающие лейкоциты (нейтрофилы), иногда макрофаги.
Цилиотоксины — факторы возбудителей респираторных инфекций, вызывающие цилиарную дискинезию, цилистаз и цилиотоксичность (например, трахеальный токсин B. pertussis, пневмолизин S. pneumoniae).
Суперантигены — неспецифические (поликлональные) стимуляторы лимфоцитов, прежде всего Т-лимфоцитов. В отличие от моноклональной (антигензависимой) активации, в реакцию вовлекается множество клонов лимфоцитов, что ведет к гиперсекреции цитокинов и синдрому вторичной (цитокинопосредованной) интоксикации. Свойствами суперантигенов обладают энтеротоксины и токсины токсического шока стафилококка, скарлатинозный токсин стрептококка, антигены микоплазм и др. Известны суперантигены, которые неспецифически стимулируют В-лимфоциты, вызывая поликлональный синтез иммуноглобулинов.
Избирательность токсического эффекта определяется спецификой клеточных рецепторов. Некоторые из токсинов воспринимаются ганглиозидами цитоплазматической мембраны (энтеротоксин холерного вибриона, нейротоксины клостридий, вызывающих столбняк и ботулизм). Т-суперантигены обретают активность после связывания с молекулами главного комплекса гистосовместимости (HLA-2) макрофагов, действие ЛПС-эндотоксинов опосредовано через рецепторы макрофагов для сывороточных белков (ЛПС-связывающий белок, производные комплемента). Для многих токсинов природа рецепторов не установлена.
По механизму действия можно выделить несколько групп токсинов:
1. Токсины, действующие на цитоплазматическую мембрану. Структурное повреждение мембран и лизис клеток-мишеней вызывают фосфолипазы (например, альфа-токсин Clostridium perfringens), детергенты (альфа-токсин Staphylococcus aureus), факторы, агрегирующие холестерол (О-стрептолизин Streptococcus pyogenes). Повреждение может быть функциональным, ограничиваясь нарушением активности мембранных ферментов. Иногда это смертельно для клетки, хотя и не сопровождается цитолизом. Например, лейкоцидин золотистого стафилококка токсичен для нейтрофилов, так как, повышая проницаемость для Са2+, дестабилизирует внутриклеточный ионный баланс.
2. Внутриклеточные токсины. Внедряются в клетку, атакуя внутриклеточные мишени. Этому обычно предшествует активация токсина — ограниченный протеолиз и восстановление внутримолекулярных тиоловых связей. При активации от молекулы отщепляется фрагмент, который проникает в клетку, оказывая каталитическое действие. Структуру таких токсинов (бинарные токсины) описывают формулой А-В, где «В» выполняет рецепторную функцию, а «А» отвечает за токсичность.
Один из универсальных каталитических эффектов — АДФ-рибозилирование ключевых ферментов клетки. Принцип сводится к отщеплению аденозиндифосфата (АДФ-рибозы) от никотинамидадениндинуклеотида (НАД) и его присоединению к белку-мишени. Полагают, что специфические ингибиторы АДФ-рибозилирования могут составить новую группу высокоактивных антагонистов микробных токсинов.
Токсины этого типа (точнее их А-фрагмент) являются бифункциональными ферментами, действуя как НАД-гидролазы и АДФ-рибозилтрансферазы. Суммарно реакция выглядит следующим образом: 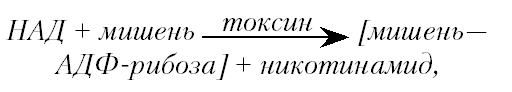

где «мишень» и «мишень—АДФ-рибоза» — функционально активный и инактивированный токсином внутриклеточный фермент.
А-фрагменты бинарных токсинов обладают еще одним важным свойством — высокой избирательностью катализа. Попав в клетку, каждый из них находит собственную мишень, модифицируя строго определенные ферменты. В сочетании со спецификой связывания В-фрагмента это объясняет особенности биологического эффекта разных токсинов. Например, дифтерийный токсин инактивирует (АДФ-рибозилирует) фактор элонгации-2, нарушая сборку пептидов на рибосомах. Энтеротоксин холерного вибриона (холероген) модифицирует белок (G-белок), блокирующий аденилатциклазу энтероцитов; это ведет к повышению внутриклеточного уровня циклического аденозинмонофосфата (цАМФ), гиперсекреции изотонической жидкости в тонком кишечнике и дегидратации организма. Кстати, известны токсины, обладающие прямой аденилатциклазной активностью (один из минорных токсинов коклюшной палочки, отечный фактор токсина сибиреязвенных бацилл). Они извращают функции клеток, нарушая внутриклеточный гомеостаз по цАМФ.
Разновидностью внутриклеточных токсинов являются нейротоксины клостридий, вызывающих столбняк и ботулизм. Их тропизм к нервной ткани определяется ганглиозидными рецепторами нейронов и нервных окончаний. При столбняке токсин (тетаноспазмин) образуется в ране и после связывания окончаниями периферических нервов достигает спинного мозга путем ретроградного внутриаксонального транспорта. В спинном мозге он воспринимается регуляторными нейронами, блокируя секрецию ингибиторных нейротрансмиттеров — глицина и гамма-аминобутировой кислоты. Это вызывает перевозбуждение моторных нейронов, приводя к стойким мышечным контрактурам — спастическим параличам. Ботулинический токсин поступает с пищей, контаминированной клостридиями. Он устойчив к протеолитическим ферментам пищеварительного тракта и после всасывания в кровь достигает нервно-мышечных синапсов, где связывается рецепторами пресинаптической мембраны двигательных нервов, блокируя секрецию ацетилхолина. Это обрывает нервные импульсы, приводя к необратимой релаксации мышц — вялым параличам (см. «Клостридии»).
3. Контактные (инъекционные) токсины. Известно около 20 эффекторных белков, действующих по контактному типу. Их спектр продолжает расширяться вследствие большого интереса ученых к функциональным последствиям адгезии.
К инъекционным токсинам можно отнести белки грамотрицательных бактерий (энтеробактерии, псевдомонады и др.), которые вводятся в клетку при адгезивных контактах благодаря механизму секреции типа III. В случае нефагоцитирующих (эпителиальных) клеток их действие может быть направлено на перестройку актинового цитоскелета, которая подготавливает поглощение бактерий (шигеллы, сальмонеллы) подобно инвазинам наружной мембраны (йерсинии). Энтеропатогенные и энтерогеморрагические эшерихии секретируют белок (Tir/EspE), который включается в плазматическую мембрану клеток и, связывая специальный адгезин (итимин), служит рецептором для усиления адгезии. Еще одним эффектом является запуск апоптозной программы клеток. Это замечено для штаммов Shigella, Yersinia, Salmonella и Pseudomonas, которые убивают фагоциты, впрыскивая в них факторы, активирующие каспазный цикл. Сальмонеллы способны усиливать свое выживание в макрофагах, продуцируя контактные токсины, блокирующие слияние фагосом и лизосом.
4. Модулины. Так называются микробные факторы, действующие опосредованно, через медиаторы воспаления и иммунитета. Это связано с тем, что при контакте с фагоцитами, лимфоцитами и другими клетками они побуждают к продукции цитокинов/хемокинов, которые отвечают за токсический эффект. Одной из молекулярных мишеней служат TLR-рецепторы клеток (от англ. toll like receptor), которые передают сигнал через систему транскрипционных факторов (прежде всего через NF-kB) на генетический аппарат клетки, стимулируя экспрессию цитокиновых генов.
Примером модулинов являются ЛПС-эндотоксины. В патогенезе эндотоксиновой интоксикации задействовано множество мишеней, но центральным звеном являются макрофаги, с которых начинается каскад ЛПС-инициируемых реакций. Напрямую ЛПС-эндотоксины слабо стимулируют фагоциты, но их активность возрастает при взаимодействии с ЛПС-связывающими белками плазмы. Главный из них принадлежит к семейству острофазовых белков и после взаимодействия с липидным фрагментом ЛПС воспринимается рецепторами CD14, которые сами не способны транслировать данный сигнал на эффекторный аппарат клетки, но добиваются этого при помощи ассоциированного с ними TLR-4. Макрофаги начинают секретировать цитокины, возбуждающие реакции новых мишеней, из которых складывается патогенез эндотоксинемии (см. «Сепсис»).
К категории модулинов принадлежат и суперантигены, о которых говорилось выше. Они действуют главным образом через иммунные рецепторы Т-лимфоцитов, минуя их эпитопную специфичность. Это вызывает поликлональную (неспецифическую) стимуляцию лимфоцитов, способствуя гиперсекреции цитокинов, которые включаются в патогенез интоксикации, а иногда и определяют ее основное содержание.
Иммунология токсинов
Все токсины — отличные иммуногены, но протективность антител лучше всего выражена в реакциях с экзотоксинами. Нейтрализовать эндотоксины труднее, возможно потому, что антигенные и токсические свойства ЛПС связаны с разными участками молекулы. В отличие от эндотоксинов, экзотоксины легко поддаются обезвреживанию без потери иммуногенности (например, формалином) и могут быть использованы в качестве вакцинных препаратов — анатоксинов. Однако антитела нейтрализуют только свободные токсины и бессильны против их каталитически активных фрагментов, проникших в клетку. Этим, в частности, объясняется малая эффективность антитоксических сывороток (антитоксинов) в лечении столбняка и ботулизма. Требуется опережающий иммунный ответ, который может быть получен лишь в условиях анамнестической (ревакцинальной) реакции.
Опосредованная болезнетворность
Микроб — ничто, субстрат — все. Согласно легенде, эти слова, подчеркивающие значимость реакций хозяина во взаимоотношениях с микроорганизмами, принадлежат Л.Пастеру. Действительно, бактерии, попадая в организм, не остаются незамеченными, вызывая реакции врожденного и адаптивного иммунитета. Это великолепно просматривается в концепции о модулинах, которые, причиняя функциональное повреждение, являются одним из факторов, способствующих воспалению в зоне внедрения возбудителя.
Исходно нацеленное на борьбу с инфектом, воспаление нередко становится патогенетической доминантой процесса, требуя лечебного вмешательства. Так, в частности, обстоит дело с заболеваниями пищеварительного, дыхательного и урогенитального трактов, которые до основной патологии начинаются с адгезии бактерий на эпителиальных клетках и быстрого выделения ими флогогенных медиаторов, прежде всего цитокинов/хемокинов. Мысль о приоритете контактной болезнетворности перед такими традиционными представлениями об эпителиотропной пиогенности, как инвазия и цитодеструктивность, имеет серьезные основания.
Что касается специфического воспаления, которое следует за иммунологической реакцией на микробные антигены, то бактериологии известен туберкулез и ряд других инфекций, формирующихся на основе иммунопатогенетических процессов (см. «Туберкулез»).
Персистенция
Длительное сожительство бактерий с хозяином — широко распространенное явление. Достаточно напомнить об огромном количестве бактериальных видов, составляющих нормальную микрофлору (см. «Нормальная микрофлора и дисбактериоз»). Но бессимптомное носительство возможно и для патогенных бактерий. Принципиально, что оно протекает на фоне приобретенного иммунитета, эффекторы которого оказываются бессильны против утвердившегося инфекта. Еще удивительнее, что для некоторых бактерий персистенция — патогенетическая норма, определяющая сущность инфекционного процесса (микобактерии, трепонемы, бруцеллы, гонококк, хламидии). Это означает, что возбудитель располагает механизмами, которые обеспечивают его выживание и даже агрессивность в условиях иммунитетного пресса.
Как уже говорилось, в ряде случаев патогенетически значимая персистенция поддерживается за счет паразитирования внутри макрофагов. Заражение эпителиоцитов, которым определяется патогенез некоторых бактериальных инфекций, не обеспечивает долгосрочного убежища, так как эпителиальные клетки быстро гибнут, возбуждая острую воспалительную реакцию.
Антигенная мимикрия. Немало спекуляций построено на феномене эпитопной идентичности антигенов бактерий и хозяина. С одной стороны, это формальный повод для ослабления иммунного ответа (антигенное родство снижает чужеродность инфекционного агента), с другой — прецедент для аутоиммунной агрессии (частичная общность эпитопов не спасает от реакции на антиген). Ни то, ни другое реально не доказано. Значение антигенной мимикрии для персистенции возможно лишь в тех случаях, когда родство затрагивает протективные антигены, но такие примеры отсутствуют.
Антигенная изменчивость. Более важной представляется изменчивость антигенов, которая защищает возбудителя от взаимодействия с преформированными антителами. Генетическая природа этого феномена лучше всего изучена для пилей гонококка, выполняющих функции адгезинов и антифагоцитарных факторов. В структуре белка, образующего пили, имеются гипервариабельные участки, которые определяют антигенные особенности гонококковых клонов. Это связано со случайной перетасовкой фрагментов ДНК, из которых складываются финальные гены пилей. Благодаря этому каждый штамм может стать родоначальником множества клонов, непохожих друг на друга по протективным антигенам. Селекция иммунорезистентных клонов является одной из причин стабилизации хронического гонорейного процесса. Интересно, что сходный механизм лежит в основе структурной вариабельности антигенсвязывающих доменов иммуноглобулинов, определяющий уникальную специфичность антител.
Изменение антигенной структуры поверхностных белков возможно в результате фенотипически значимых переключений генотипа, известных как фазовые вариации. В этом случае бактерии обладают регулятором транскрипции, который ответственен за включение одной группы генов и выключение других групп. Это тоже совершается стихийно, но возникающие антигенные варианты используются для ускользания от преформированных антител. Механизм фазовой изменчивости выражен у сальмонелл, возбудителя коклюша, нейссерий.
Есть и более простой способ уклонения от эффекторов иммунитета. Некоторые бактерии фиксируют на своей поверхности белки хозяина, прикрывая ими собственные антигены. Мы уже говорили о фибриновой псевдокапсуле стафилококка. Нечто подобное образует возбудитель сифилиса, сорбируя b-2-микроглобулин и/или тканевой мукополисахарид. Хорошо известна бактериотропность фибронектина (например, в отношении стафилококков и трепонем), но ее влияние на взаимоотношения с хозяином остается неясным.
L-формы. Все бактерии, за исключением микоплазм, обладают жесткой оболочкой (клеточной стенкой), которая, подобно покрышке надувного мяча, окружает цитопласт, придавая клеткам определенную форму. У большинства бактерий механическая прочность клеточной стенки зависит от ее главного структурного компонента — пептидогликана, или муреина (см. «Антибиотики»). Он выдерживает большое осмотическое давление (у грамположительных бактерий до 30 атм), стремящееся разорвать хрупкую плазматическую мембрану изнутри. Повреждение пептидогликана (например, лизоцимом) или нарушение его синтеза (бета-лактамные антибиотики) ведет к гибели бактерий, и, казалось бы, не требует комментария.
Но дело обстоит сложнее, так как некоторые из бактериальных клеток не только выживают без стенки (протопласты, сферопласты), но и сохраняют способность к размножению. Они получили название «L-форм» в честь Дж. Листера (первое описание L-форм сделано E. Klieneberger в 1936 г. в институте им. Листера, Англия). E. Klieneberger обнаружила их в культуре Streptobacillus moniliformis (возбудитель лихорадки крысиных укусов), расценив как неизвестный ранее симбионт бактерий. После того, как L.Dienes удалось вернуть L-колонии в обычную форму, стало ясно, что речь идет не о новой группе микроорганизмов, а о своеобразной изменчивости бактерий, связанной с полной или частичной потерей клеточной стенки. Интерес к этому явлению породил множество исследований, нацеленных на выяснение биологической (в том числе патогенетической) сущности феномена L-трансформации. Пик этих исследований приходится на 1960—1980 гг.
Кроме возможности обратной трансформации (реверсии) в бактериальные формы, в поддержку болезнетворности L-форм приводились факты о сохранении у них функции экзо- и эндотоксинообразования, способности продуцировать ферменты патогенности, повышенной резистентности к антибиотикам (они нечувствительны к бета-лактамным антибиотикам), антителам и комплементу (за счет утраты многих поверхностных антигенов). Много внимания уделяется персистенции L-форм, которая способствует длительному выживанию ряда патогенных бактерий в организме человека и животных (туберкулезная палочка, стрептококки, гонококки, листерии, бруцеллы, трепонемы и пр.). В этом видится экологический аспект проблемы — сохранение паразитарных бактерий в природе за счет пусть болезненной для них (патологической), но все-таки поддерживающей жизнь адаптации.
Гораздо меньше согласия у ученых в признании патогенетического потенциала L-персистенции. Несмотря на то, что ряд авторов поддерживает участие L-форм в развитии этиологически нерасшифрованных, хронических заболеваний (аутоиммунные процессы, ревматизм и гломерулонефрит, болезнь Крона и пр.), многие считают это недоказанным.
Одним из поводов к сомнениям является хрупкость L-форм. Для их выживания требуются гипертонические питательные среды, уравновешивающие внутриклеточное осмотическое давление (этого добиваются при помощи сред с высокой концентрацией поваренной соли или сахарозы). Но известны и соленезависимые варианты (те же L-формы S. moniliformis), которые «самостабилизируются» за счет системы внутренних мембран, отсутствующей у бактериальных форм. Следует помнить и о том, что между бактериями с полноценной клеточной стенкой и вариантами, лишенными пептидогликана, есть переходные (промежуточные) формы с различной выраженностью дефекта.
Есть мнение, что конверсия в L-формы достаточно распространена в природе как один из этапов циклического развития бактерий. C этой точки зрения для индукции L-форм вовсе не обязательны искусственные манипуляции, побуждающие сомневаться в реальности феномена in vivo. Уже давно, например, сообщалось о том, что тот же пенициллин не столько индуцирует, сколько селекционирует спонтанно образовавшиеся L-клетки стрептококков. Фактология о дремлющих дефектных бактериях недавно получила серьезное методическое подспорье в виде полимеразной цепной реакции, позволяющей улавливать контаминацию тканей даже в тех случаях, когда возбудитель не обнаруживается традиционными способами (см. «Диагностика инфекционных заболеваний»).
В 1985 г., критически оценивая проблему, С.В. Прозоровский резюмировал: «На смену большим ожиданиям пришел жесткий анализ фактов. L-формы могут иметь значение далеко не при всех инфекционных процессах. Наиболее вероятна их роль в развитии хронических и рецидивирующих инфекций. Система доказательств представляется достаточно сложной, она требует дальнейших исследований и новых фактов».

Свидетельством того, что проблема жива, продолжая интриговать патологов и микробиологов, является аналитический обзор новых идей и фактов по поводу патогенетического значения L-персистенции, опубликованный в одном из журналов Американской ассоциации микробиологов2, где, в частности, говорится о целесообразности более широкого применения антимикробных средств при идиопатических, хронических и травмирующих процессах несмотря на отсутствие классических (культуральных) доказательств их бактериальной природы. Использование новых микроскопических и генетических маркеров в ряде случаев позволяет думать о присутствии гипотетического возбудителя, оправдывая применение этиотропных препаратов (см. рисунок).
Микробные сообщества (биопленки). В широком смысле к ним относятся любые агрегаты бактерий на интерфазных (вода-субстрат) поверхностях — от диффузного монослоя до массивных слизистых структур, видимых невооруженным глазом. Зрелая биопленка (англ. biofilm) представляет собой микробные образования в органическом матриксе (гликокаликсе), который включает белковые дериваты и протеогликаны хозяина. Они готовят адгезию бактерий. Биопленки являются одной из причин бактериальной персистенции, содействуя общению между микробами одного или разных видов. Они могут быть разделены каналами для поступления питательных веществ и выведения отходов. Бактерии имеют здесь разную активность, выполняя функции жизнеобеспечения всего сообщества. Не случайно биопленки называют «городами микробов», сравнивая с тканями многоклеточных организмов.
Формирование биопленки — многоступенчатый процесс. Он включает адгезию планктонных (взвешенных) клеток, закрепление адгезивных контактов и развитие многослойной структуры внутри гликокаликса. Это ведет к изменению бактериального фенотипа с индукцией множества генов, не работающих в планктонном состоянии. Бактериальные клетки периодически покидают биопленку, создавая возможность обострений и новых очагов инфекции. Переход в планктонное состояние контролируется феромонами — молекулами межклеточного общения. Используя их аналоги, удается предотвратить формирование биопленки, сдвигая баланс в сторону планктонного статуса. Это повышает чувствительность бактерий к антибиотикам и эффекторам иммунитета, помогая лечению и профилактике персистентных инфекций.
В составе биопленок бактерии имеют ряд преимуществ, к которым относятся способность к выживанию и экспрессия генов вирулентности. Во-первых, окруженные гликокаликсом бактерии недоступны для эффекторов иммунитета. Поэтому биопленочное воспаление характеризуется низкой результативностью, обретая хронический характер. Реакции хозяина в лучшем случае неэффективны, а в худшем — сопряжены с повреждением собственных тканей. Не случайно для подавления биопленочных сообществ некоторые клиницисты рекомендуют назначение иммунодепрессантов. Во-вторых, биопленки обладают высокой резистентностью к антибиотикам: она в среднем в 1000 раз выше, чем у планктонных вариантов. Кроме низкой проницаемости, это обусловлено также метаболическими особенностями биопленочных бактерий. Часть из них пребывают здесь в малоактивном (даже инертном) состоянии, а потому индифферентны к антибиотикам. Внутри биопленок поддерживаются условия для генетического обмена, в том числе для приобретения генов резистентности к антибиотикам.
Биопленочная патология. Биопленочные инфекции можно разделить на две группы — процессы, происходящие в собственных тканях хозяина, и инфицирование инородных предметов, вводимых в качестве протезов, катетеров и пр. Классическим примером патологий первого типа является зубная бляшка. В зрелой бляшке насчитывается несколько сот видов бактерий. Многие из них не удается культивировать, и об их присутствии судят по генетическим маркерам. Зубные бляшки служат причиной кариеса и пародонтита. При кариесе зубная эмаль разрушается органическими кислотами, которые образуются при сбраживании углеводов бактериями, прежде всего оральными стрептококками. Пародонтит формируется при распространении бляшки в субгингивальное пространство, где имеются условия для интенсивного размножения анаэробных бактерий с выраженным деструктивным и провоспалительным потенциалом (порфиромонады, превотеллы, фузобактерии). Хронизация процесса ведет к разрушению периодонтальных тканей и выпадению зубов.
Зубная бляшка — отличная модель, демонстрирующая фазы и механизмы развития бактериальной биопленки. События начинаются с подготовки зубной поверхности саливарными белками. Это обеспечивает адгезию и размножение первой партии бактерий — стрептококков и актиномицетов. Они продуцируют внеклеточные полисахариды (декстраны, леваны), образуя раннюю биопленку. К ранней биопленке прикрепляются другие виды бактерий, из которых формируется зрелая зубная бляшка — многослойная (толщиной до 100 мкм) полимикробная структура, скрепленная микробными и саливарными полимерами. Стабильность зубных бляшек (для их удаления необходима интенсивная чистка зубов) отличает их от экологии слизистых оболочек, которые обновляются каждые несколько дней. При слущивании мукозального эпителия удаляются микроорганизмы, оставляющие потомство на новых клетках. Поэтому микробные сообщества нормальных слизистых оболочек являются монослойными, не достигая организации зрелых биопленок. Тем не менее известны заболевания, связанные с образованием микробных сообществ на мукозальных поверхностях. Полагают, что около 60% бактерийных инфекций так или иначе связаны с биопленками или сходными с ними образованиями. К наиболее типичным патологиям относятся септический эндокардит и муковисцидоз, хотя в любой хронической инфекции можно подозревать участие биопленочного процесса.
Септический эндокардит объединяет хронические бактериальные инфекции клапанов сердца. В типичных случаях бактерии колонизируют тромбоподобные массы, которые откладываются при бактериемии на поврежденном эндотелии (ревматический порок, хирургические вмешательства). Источником бактериемии обычно служит ротовая полость, реже — интестинальный и урогенитальный тракты. Этим определяется микробный профиль эндокардитов, в котором преобладают оральные стрептококки; далее следуют стафилококки и грамотрицательные бактерии. Нередко инфекция носит смешанный характер.
Бактерии прикрепляются к элементам тромба, образуя микроколонии, которые покрываются новыми тромбоотложениями. Повторение циклов ведет к развитию многослойных биопленочных структур — так называемых вегетаций. Они служат источником септической интоксикации, а также причиной органических повреждений клапанов сердца и миокарда, тромбоэмболий. При дентальных или других инвазивных операциях больным с повышенным риском эндокардита рекомендовано назначение антибиотиков. Это делается для того, чтобы убить планктонные бактерии в сосудистом русле, прежде чем они сформируют биопленочное сообщество на клапанах сердца.
Муковисцидоз принадлежит к числу наиболее распространенных врожденных патологий. В 70% случаев причиной служит генетический дефект по белку, регулирующему секрецию воды мукозальными эпителиоцитами. Подавление мукоцилиарного транспорта снижает эффективность самоочищения бронхопульмонарного тракта, создавая условия для микробной колонизации. В финальной стадии доминируют мукоидные штаммы синегнойной палочки (см. «Синегнойная палочка»). Они образуют биопленку, внутри которой бактерии проявляют патогенетическую активность и ограждены от эффекторов иммунитета и антибиотиков.
Проблема биопленок впервые возникла в технической микробиологии в связи с громадным экономическим ущербом, наносимым микробным обрастанием разного рода водных сооружений. В медицине сходные ситуации обусловлены использованием инвазивных материалов — протезов, катетеров, эндотрахеальных трубок, артериовенозных шунтов, контактных линз, внутриматочных спиралей и пр. Они почти неизбежно подвергаются колонизации экзогенными и/или эндогенными бактериями, образующими на них стабильные сообщества. Образование биопленок на эндоваскулярных протезах (клапаны сердца, сосуды), внутрисосудистых и интраперитонеальных катетерах (хронический диализ) начинается с заселения эпидермальным стафилококком (см. «Стафилококки»). Позже процесс обретает полимикробный характер за счет добавления зеленящих стрептококков, Staphylococcus aureus, Pseudomonas aeruginosa, Enterococcus faecalis, Candida albicans и др. При катетеризации мочевыводящей системы в состав биопленок входят бактерии, утилизирующие мочевину (Proteus mirabilis, Proteus vulgaris, Klebsiella pneumoniae, Morganella morganii), хотя первичную колонизацию могут вызывать другие бактериальные виды (Staphylococcus epidermidis, Escherichia coli, Enterococcus faecalis).
К счастью, ятрогенные процессы обычно малоагрессивны. Это объясняется низкой вирулентностью бактерий, которые в своем большинстве представлены нормальной микрофлорой. Впрочем, подобно любой персистенции биопленочная инфекция таит в себе потенциальную опасность, требуя адекватного контроля. Природа сама стремится к этому: инородные тела подвергаются эпителизации, а эпителиальная поверхность гораздо устойчивее к микробному обсеменению, чем синтетические материалы. От такого рода конкуренции за поверхность во многом зависит стабилизация микробных биопленок.
Генетические основы болезнетворности
Реализация патогенного потенциала
Как и любые клетки, бактерии экспрессируют лишь часть фенотипически значимых генов. Среди них есть такие, которые детерминируют обязательные признаки — конститутивные гены (англ. house-hold genes). Они обеспечивают видовой облик бактерий, т.е. их базисную конституцию — морфологию, тинкториальные свойства, метаболический профиль и пр. Кроме того, ряд генов включается по мере необходимости, в зависимости от обстановки, куда попадают бактерии. Это индуцибельные гены, кодирующие необязательные признаки. Речь, по сути, идет об экологически зависимой экспрессии генетического материала, которая обеспечивает пластичность бактерий, т.е. их адаптивный потенциал. Например, у энтеробактерий из 3000 генов примерно треть лишена фенотипического эквивалента. То же самое справедливо и для других бактерий.
Современное понятие о функциональной генетике предполагает изучение всего спектра белков (его называют протеом), которые закодированы в геноме и могут синтезироваться клеткой. Это связано с механизмами, обеспечивающими активацию или блокаду индуцибельных генов (генов адаптации). Классическим примером является оперон. Это одиночный ген или группа генов, координируемых одним оператором — механизмом включения промоторного участка структурных генов. Активность гена-оператора контролируется геном-регулятором, который кодирует белок-репрессор, или белок-регулятор. Имея сродство к гену-оператору, регулятор блокирует его промоторную активность, выключая транскрипцию структурных генов. Вместе с тем, регуляторы специфически связывают низкомолекулярные вещества, называемые индукторами. Соединение с таким индуктором меняет конформацию регулятора, лишая его способности связываться с геном-оператором. Выходя из-под контроля, оператор обеспечивает синтез мРНК и соответствующего белка.
Несколько оперонов (генных кластеров), управляемых общим фактором, составляют регулон. Регулоны определяет возможность коэкспрессии (корегуляции) генов, в частности, генов, отвечающих за вирулентность. В других ситуациях каждый из таких генов может экпрессироваться независимо, реагируя на дискретные сигналы.
Системы регуляции бактериального фенотипа разнообразны, но в целом не оригинальны, используя унифицированные механизмы детерминации физиологических процессов в клетке. В конечном счете все сводится к управлению транскрипцией генов. Одними из универсальных являются двухкомпонентные (двухсигнальные) регуляторные системы. Они повторяют общий принцип экологически зависимой регуляции генов, когда сигналы из внешней среды улавливаются сенсорными белками (рецепторами) и затем транслируются на белки-регуляторы (посредники между сенсорами и генами), активируя или репрессируя транскрипцию генов. Изменение функционального статуса сенсорных и регуляторных белков часто связано с их фосфорилированием (в первом случае аутофосфорилирование, во втором — результат действия киназ, ассоциированных с сенсорными рецепторами). Перенос фосфатного остатка на аминогруппу белка-регулятора нарушает его способность к связыванию с ДНК-последовательностями, с которого начинается транскрипция оперона (регулона).
Реализация болезнетворности бактерий зависит и от синтеза новых регуляторных факторов. Их предложено называть стресс-индуцибельными белками, так как они образуются при реакции на неблагоприятные или, по крайней мере, необычные условия, в том числе возникающие в инфицированном организме. Бактерии хорошо чувствуют перепады температуры, напряжение кислорода, рН среды, содержание кальция, дефицит питательных веществ и пр. Попадая в организм хозяина (или выходя из него), они сталкиваются с массой новых факторов, требующих адаптивных реакций. Это особенно заметно у бактерий, чередующих сапрофитический и симбиотический образ жизни, но актуально и для облигатных паразитов. Примером является s(сигма)-регулон энтеробактерий. s-фактор (его образование связано с индукцией rpoS/katF-гена) регулирует транскрипцию многих генов, включая гены, детерминирующие осмопротекцию, морфологию клеток, выживание в условиях голодания и пр. Известны и другие механизмы регуляции генов, основанные на изменении конформации репрессорных белков, топологии ДНК и пр. Все они связаны с реактивной перестройкой бактериальных клеток под влиянием факторов внешней среды.
Адаптивные проявления изменчивости бактерий необозримы. Классикой являются экологически зависимое чередование споровых и вегетативных форм, интенсивность образования капсульного материала, феномен диссоциации культуральных свойств (в частности, S-R-диссоциация), синтез индуцибельных ферментов и т.д. Известны примеры, связанные с формированием вирулентного фенотипа. Холерные вибрионы, попав из сапрофитической фазы в кишечник человека, коэкспрессируют гены, детерминирующие синтез адгезинов и энтеротоксина (индуцирующее действие контакта с энтероцитами, температуры тела и пр.). Только в такой комбинации холерные вибрионы могут вызывать заболевание. Сальмонеллы обладают адаптацией (толерантностью) к кислой среде, усиленно экскретируя протоны из клеток. Оказавшись внутри макрофагов, сальмонеллы экспрессируют около 200 генов, которые не работают при внеклеточном росте. Их продукты обеспечивают не только выживание, но и активное размножение внутри фагоцитов. То же самое справедливо для других бактерий, склонных к внутриклеточному паразитированию (йерсинии, листерии, шигеллы и пр.). Шигеллы по ходу инфекционного процесса теряют толерантность к кислой среде, но к моменту выхода во внешнюю среду вновь обретают ее, словно готовясь к очередному сеансу заражения и преодолению кислотного барьера желудка. Этим определяется минимальная среди всех бактерий инфицирующая доза шигелл (менее 1000 клеток). Коклюшная палочка, попадая в бронхи, начинает продуцировать комплекс токсинов, но, подвергаясь выселению в носовые ходы, где температура гораздо ниже, трансформируется в невирулентный фенотип. Легко меняют свои свойства йерсинии, в частности Y. enterocolitica. Развиваясь при 37°С, они продуцируют белки наружной мембраны, сидерофоры, ферменты, которые повышают колонизирующую активность бактерий, усиливают их инвазивность, устойчивость к комплементу и фагоцитозу.
Адаптивный смысл повышенной вирулентности можно объяснить тем, что, обладая высокой способностью к размножению в организме хозяина, бактерии более эффективно утверждают себя в биосфере. Это прежде всего относится к микробам с узким спектром хозяев (например, возбудителям антропонозов), которые при активном размножении получают возможность для трансмиссии, т.е. заражения новых организмов.
Большой резонанс получило изучение контактных взаимодействий бактерий с клетками хозяина. Эти взаимодействия лежат в основе патогенетически значимых взаимоотношений с мукозальными эпителиоцитами. Адгезивные контакты, реализуемые через комплементарные рецепторы бактерий и клеток, инициируют сигналы, оказывающие серьезное влияние на функциональное состояние каждого из взаимодействующих сочленов. Результатом является последовательный переход от рыхлых (фимбриальных) контактов к интимной адгезиии, построение специализированных органелл секреции (аппарат секреции III типа), синтез и направленный транспорт в клетки инъекционных токсинов. Экспрессия индуцибельных генов проявляется здесь в виде контактной патогенности — одного из универсальных механизмов реализации болезнетворности бактерий.
В целом ясно, что инвитровые культуры дают неполное представление о факторах микробной болезнетворности, так как некоторые из признаков, определяющих вирулентность бактерий, могут проявиться только внутри чувствительного организма. Поэтому вопрос «что же такое патогенность?» до сих пор во многом риторичен. Современные методы позволяют судить об особенностях экспрессии микробных генов в различных ситуациях и об их участии в экологически зависимой адаптации бактерий. Парадокс, но, регистрируя индуцибельные гены, задействованные в реализации вирулентности, мы далеко не всегда знаем их продукты. Изучением этого вопроса занимается функциональная генетика, одной из задач которой является стыковка генома с протеомом, т.е. расшифровка фенотипического выражения всего генетического материала клетки.
Эволюция болезнетворности
Эволюция живых организмов связана с изменением генетического материала и селекцией его оптимальных, т.е. наиболее приспособленных к данной среде вариантов. Патогенные микроорганизмы явились результатом развития биоагрессивных генотипов. Одновременно формировались механизмы их регуляции, связанные с экологически зависимой экспрессией генов вирулентности.
Поскольку прокариоты были первой формой жизни на Земле, бактериальные симбионты эволюционировали на основе сапрофитов. Эксплуатируя хозяина, они утратили часть признаков свободноживущих форм, но одновременно обрели гены, детерминирующие агрессивность. Адаптивный смысл болезнетворности не совсем понятен. Более того, она похожа на парадокс, не выгодный паразиту. Гораздо логичнее — спокойное сожительство, когда симбионт получает возможность для бесконфликтного, а потому надежного существования. С этой точки зрения, патогенность выглядит как результат случайного стечения обстоятельств, а инфекционные болезни — как необязательное условие для развития микроорганизмов. Но ситуация может быть и иной, когда инфекционный конфликт обретает позитивное (для возбудителя) значение. Усиленное размножение создает условия для совершенствования бактерий на основе селекции клонов с обновленным генотипом. Это реально не только для антропонозов (когда человек служит единственным резервуаром инфекции), но и для возбудителей, которые способны к автономному существованию во внешней среде (сапронозы) или к заражению других хозяев (зоонозы). Если учесть, что большинство патогенных микроорганизмов имеют экологические ниши, где они бесконфликтно персистируют, то заражение чувствительных организмов служит своеобразным допингом, создавая дополнительные возможности для селекции и, следовательно, эволюции.
Следует помнить и о том, что многие патогенные виды клонированы (дифференцировку клонотипов проводят на основе генотипирования — «генетические отпечатки пальцев», англ. genetic fingerprinting), и лишь часть (нередко малая) клонов обладают агрессивностью. Например, из 100 клоногрупп менингококка группы А эпидемическое значение имеют не более десятка клонотипов. Среди более 100 клонотипов палочки инфлюэнцы типа b всего 6 отвечают за 80% наиболее серьезных инфекций. Это означает, что даже самые грозные патогены способны существовать на основе компромисса с хозяином, а болезнетворность может быть скорее исключением, чем правилом, показывая, как мало надо иметь, чтобы обрести вирулентность или утратить ее. Для большинства патогенных бактерий известны двойники или близкие виды, которые лишены болезнетворности, но тем не менее великолепно существуют в природе как сапрофиты или симбионты. Обретение вирулентности скорее усложнило, чем упростило их жизнь.
Бактериальные гены оформлены в виде двух базисных структур — хромосомы и плазмид. Это самостоятельные репликоны, т.е. геномные молекулы, способные к саморазмножению (репликации). Хромосома включает около 3000 генов, большинство из которых кодирует жизненно важные функции бактерий. Плазмиды (их называют минихромосомами) включают 20—200 генов. Многие из них имеют адаптивное значение, проявляются в неблагоприятных или конкурентных условиях (например, устойчивость к антибиотикам, продукция иммунитетных факторов и пр.). Поэтому плазмиды можно элиминировать, сохранив жизненный потенциал их хозяев.
Кроме мутаций, у хромосом и плазмид есть еще одно предназначение: они служат местом внедрения (инсерции/интеграции) новых генов, обладающих способностью к транспозиции. Перемещаясь из клетки в клетку, из репликона в репликон, такие гены эволюционируют как рекомбинантные молекулы и поэтому называются транспозонами. Их способность включаться в состав репликонов отражена понятием интегрон. Cамые простые транспозоны обозначаются как вставочные гены, или IS-элементы (от англ. insertion sequence — вставочная последовательность). Они лишены экзонов, т.е. генов, кодирующих фенотипические признаки. Единственное, что умеют делать IS-транспозоны — это включаться и выходить из состава репликонов. Их влияние на фенотип связано с инсерционным мутагенезом, т.е. с нарушением структуры (непрерывности) генов, подвергшихся инсерции/вставке. В транспозиции могут участвовать бактериофаги, воспринимающие чужие транспозоны или фрагменты бактериальных репликонов (хромосома, плазмиды). Генетическому обмену содействуют микробные сообщества типа биопленок, где формируются плотные контакты на основе клеточных агрегатов из разных видов бактерий.
В отличие от высших организмов, для которых единственным механизмом генотипической изменчивости является вертикальный (от родителей потомству) путь передачи обновленных комбинаций аллельных генов, горизонтальная или латеральная трансмиссия генов у бактерий играет решающую роль. Таким путем передаются хромосомные гены и плазмиды, транспозоны и фаговые геномы. Наиболее известными механизмами являются трансформация (пассивное проникновение ДНК в бактериальные клетки), трансдукция (мобилизация генов при помощи фагов) и конъюгация (физический контакт бактерий, детерминируемый плазмидами). Горизонтальная передача существенно расширяет экологическую пластичность бактерий, позволяя использовать эволюционный опыт родственных и таксономически отдаленных видов. Иногда это чревато осложнениями для биосферы, порождая потенциально опасные генотипы.
Включение чужих фрагментов ДНК в состав собственных репликонов бактерий происходит благодаря негомологичной (незаконной) рекомбинации, основанной на точечной (сайт-специфической) комплементарности взаимодействующих молекул. Этим негомологичная рекомбинация отличается от гомологичной, которая совершается внутри половой зиготы при полном взаимодействии (кроссинговере) аллельных генов хромосом одного вида. Это объясняет присутствие однотипных генов у бактерий разных таксономических групп. Некоторые из них имеют отношение к болезнетворности, формируя генные кластеры, известные как острова патогенности. В их функции, в частности, входит реализация контактной болезнетворности, т.е. образование адгезинов и секреция контактных/инъекционных токсинов. Острова патогенности могли быть внесены бактериофагами или плазмидами и включены в состав бактериальных хромосом путем инсерции негомологичных интегронов.
Формирование островов патогенности происходило скорее всего неодновременно. Они отражают результат последовательных вставок, потребовавших значительного времени и уникального стечения обстоятельств. Консервативность вирулентных генов отражает общность связанной с ними стратегии и их единой природы. Впрочем, это лишь та основа, которая в ходе многовековой эволюции подверглась существенным изменениям и дополнениям. Острова патогенности обнаружены далеко не у всех болезнетворных бактерий. Они характерны для грамотрицательных бактерий и лучше всего изучены у патогенных энтеробактерий (сальмонеллы, шигеллы, диареегенные эшерихии, иерсинии).
*** Завершая обзор, посвященный факторам и механизмам болезнетворности бактерий, мы не закрываем тему. Ее мотивы пройдут через все очерки, связанные с проблемами частой бактериологии.
Болезнетворность вирусов
- Эволюция болезнетворности.
- Деструкция клеток.
- Образование симпласта.
- Апоптоз.
- Онкогенная трансформация.
- Иммунопатогенез вирусных инфекций.
- Персистенция.
- Медленные инфекции и прионы.
Вирусная инфекция эволюционирует на уровне чувствительных (пермиссивных) клеток, куда вирус вносит свои гены. От их экспрессии зависят судьба клетки, вируса и клинические проявления инфекционного процесса. Селекция поражаемых клеток начинается с распознавания клеточных рецепторов, комплементарных адгезивным белкам вириона. Это означает, что каждый вирус находит собственную молекулярную мишень, которая выводит его на определенную категорию пермиссивных клеток. Некоторые (возможно, многие) вирусы распознают несколько мембранных структур, и связывание представляет собой многоступенчатый (в том числе с функциональной точки зрения) процесс (см. «ВИЧ-вирусы»). После проникновения в клетку вирус (точнее, вирион) распадается на субкомпоненты с высвобождением геномной молекулы (ДНК или РНК). Она определяет дальнейшую стратегию вируса — репродукцию новых вирусных частиц (продуктивная, или репликативная инфекция) или персистенцию с более или менее ограниченной экспрессией вирусных генов (вирогения). И то и другое чревато повреждением клеток — цитопатическим эффектом.
Повреждение означает комплекс структурно-функциональных нарушений, которые заканчиваются гибелью клетки или (если они обратимы) допускают восстановление. Иными словами, подвергаясь вирусной атаке, клетки, подобно организму, начинают болеть, причем исходы этой болезни могут быть разными — гибель, выздоровление, уродства. К уродствам, например, относятся опухолевая (онкогенная) трансформация клеток и образование поликарионов в результате слияния нескольких клеток. Но изменения не всегда столь очевидны. Они могут ограничиться появлением на поверхности зараженных клеток новых (вирусных) антигенов, в том числе в комплексе с HLA. Впрочем, и такая перестройка может быть значимой, превращая клетку в мишень для иммунных реакций. На этом основано выделение двух механизмов вирусной цитопатогенности — прямые и опосредованные эффекты. Первые целиком зависят от самого вируса. Они отражают его взаимоотношения с клетками и могут быть воспроизведены в инвитровых культурах, т.е. вне организма. Вторые требуют участия эффекторов иммунитета и проявляются на уровне организма.
Эволюция болезнетворности
Последние годы преподнесли немало микробиологических сюрпризов, одним из которых явилось осознание того, что мы еще плохо знаем мир микробов и те опасности, которыми они угрожают человеку. Появились два англоязычных термина, emergent и reemergent infections, которые можно перевести как новые и возрождающиеся инфекции. Среди них есть заболевания вирусной природы — глобального (ВИЧ-инфекция, гепатит С) и регионального (природно-очаговые зоонозы) масштаба. Но большинство такого рода инфекций не являются новыми. Их возбудителями служат давно сформировавшиеся вирусы, с которыми люди до недавних пор встречались редко. В основе современных вспышек лежат экологические причины, связанные с активным внедрением человека в природу, когда ему приходится сталкиваться с естественными резервуарами новых для себя вирусов, таких как арбовирусы (англ. arthropod-born viruses — вирусы, передающиеся членистоногими) и родентвирусы (англ. rodent-born viruses — вирусы, передающиеся от грызунов). Имеет значение и попадание неиммунных лиц в эндемичные зоны, где благодаря широкой циркуляции возбудителя у местного населения поддерживается приобретенный иммунитет.
Вирусы подвержены генетическим изменениям на основе мутаций и рекомбинаций. Особенно быстро меняются некоторые РНК-вирусы. Это связано с тем, что клетки не контролируют точность операций, совершаемых РНК-полимеразами, как это делается в случае ДНК-полимераз. У ретровирусов к этому добавляются ошибки обратной транскриптазы, многократно усиливающие мутагенез. Подсчитано, например, что шестилетняя персистенция ВИЧ-1 в организме дает начало такому количеству вариантов, какое в течение данного времени способна обеспечить многомиллионная эпидемия гриппа А.
Большинство мутаций летальны (т.е. ведут к появлению нежизнеспособных фенотипов), но возможны и потенциально полезные перестройки, создающие прецедент для эволюции, в том числе для повышенной вирулентности в пермиссивных системах. Это зависит от естественного отбора на уровне инфицированной клетки и организма, а также при передислокации (трансмиссии) вируса очередному хозяину.
Вирусы являются результатом длительной коэволюции со своими естественными хозяевами. Переход в новую обстановку невыгоден и может обернуться катастрофой для вируса и его нового хозяина. Именно так обстоит дело с большинством зоонозных вирусов, для которых человек является экологическим тупиком. Требуется стечение многих обстоятельств, чтобы селекция в подобных условиях закончилась удачно для вируса. Очевидно, это должен быть генетический вариант с принципиально новыми свойствами, которые способны инициировать новую стратегию, предусматривающую не только внутривидовые вариации, но и видообразование. Это требует времени, которое даже у столь быстро размножающихся и мутирующих организмов, как вирусы и бактерии, несопоставимо длиннее человеческой жизни. Поэтому вирусы, которые сегодня воспринимаются как новые, в лучшем случае являются обновленными.
Одним из немногих примеров среди современных вирусов, претендующих на новый (видовой) таксономический ранг, является вирус иммунодефицита, ВИЧ-1. Его следы уходят в Африку, в 1950-е гг. Именно в это время в сыворотках аборигенов стали фиксироваться анти-ВИЧ-антитела. Тогда это была эндемичная инфекция (нечто похожее на нынешний ВИЧ-2), и причины, которые вывели ее на глобальный уровень, точно неизвестны. Скорее всего, они связаны с рекомбинантными клонами, которые возникли при инфицировании человека обезьяньим (шимпанзе) вирусом иммунодефицита. Именно от него берет начало современная генеалогия ВИЧ-1.
Внешние проявления и механизмы цитопатического эффекта
Цитолиз (некроз). В его основе лежит повышение проницаемости плазматической мембраны. Открывая доступ Са2+, это ведет к внутриклеточному ионному дисбалансу, пассивному вхождению воды, разбуханию клетки, разрыву мембраны и вытеканию цитоплазмы. В клеточных культурах этому соответствуют зоны так называемых негативных колоний — бляшек (рис. 1). Гибели клеток предшествует появление цитоплазматических или ядерных включений, которые возникают в местах сборки вирусных нуклеокапсидов и иногда заметны под микроскопом (рис. 2).



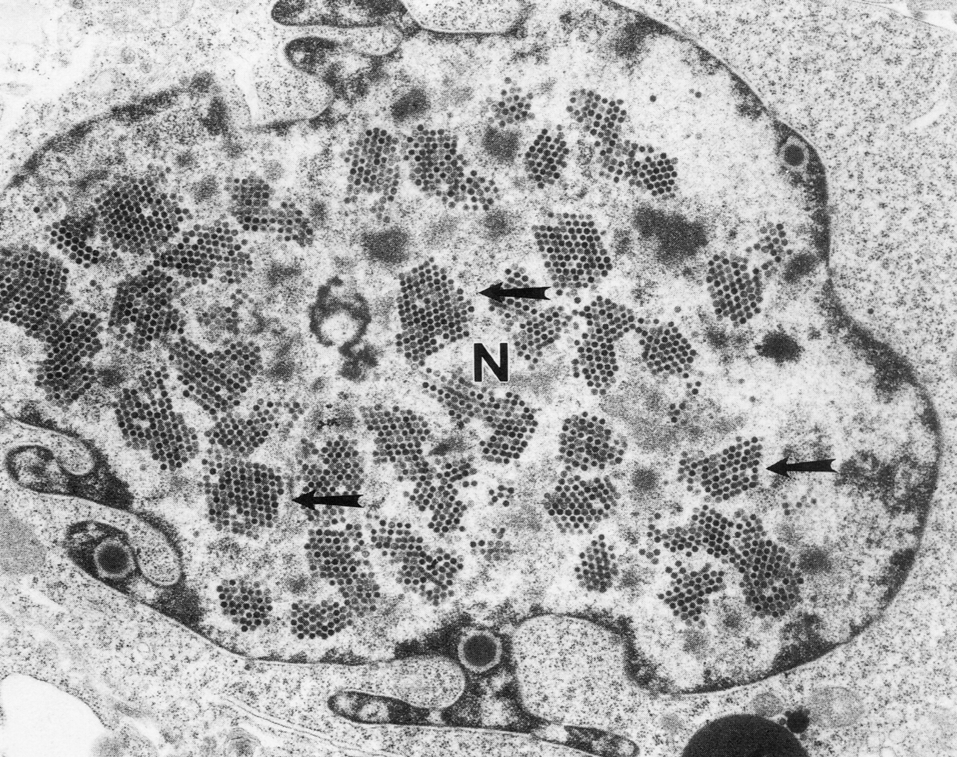
Формально цитолиз можно представить как взрыв изнутри, когда плазматическая мембрана лопается под напором вирусных частиц, скапливающихся в клетке. Высвобождение оболочечных (сложных) вирусов происходит путем почкования. В этом случае вирусный нуклеокапсид покрывается наружной оболочкой (суперкапсидом), отбирая у клетки фрагмент модифицированной мембраны. Здесь возможен феномен решета: при множественном почковании клетка не успевает ликвидировать «дыры» в мембране и предупредить вытекание цитоплазмы. Допускается и вероятность аутолиза в связи с повреждением лизосом и высвобождением из них деструктивных ферментов.
Но все это выглядит слишком прямолинейно. Клетке отдается роль пассивного полигона, где вирус беспрепятственно воспроизводит свое потомство, не встречая сопротивления. События, которые предшествуют и, возможно, способствуют цитолизу, остаются за кадром. Между тем, количество зрелых вирионов, накапливающихся в клетках, не всегда коррелирует с литическим эффектом. Известны примеры, когда клетка гибнет, не дождавшись гиперпродукции вируса. Это означает, что должны быть иные (возможно, главные) причины цитолиза, связанные с более тонким вмешательством вируса в жизнь клетки.
Вирусная инфекция — яркий пример молекулярной патологии, которая формируется под влиянием конкурентного вмешательства вирусных белков в жизненно важные процессы клетки. Жить — значит непрерывно обновлять свои функциональные и структурные элементы. Клетка, лишенная такой возможности, погибает.
Отсюда наиболее общим условием цитопатического эффекта следует считать подавление обновляющего синтеза макромолекул, стабилизирующих клеточный гомеостаз. Вирусологи называют это дискриминацией, подразумевая, что вирус вынуждает клетку переключиться на синтез своих нуклеиновых кислот и белков; самообновление клетки прекращается или задерживается. За этим скрывается множество механизмов, большинство из которых не раскрыты, хотя ряд примеров убеждает в их удивительной изящности. Так, вирус полиомиелита блокирует белок, необходимый для взаимодействия клеточных мРНК с рибосомами (кэп-связывающий белок); полиовирусная мРНК не нуждается в этой реакции, получая возможность для селективной трансляции. Действие вируса бешенства на нейроны определяется поверхностным гликопротеином G. Секвенирование (анализ нуклеотидной последовательности) клонированного гена G двух аттенуированных авирулентных штаммов показало, что лишение вируса нейротропности связано с заменой единственной аминокислоты в одном из участков гликопротеина G. Не ясно, чем обусловлен механизм аттенуации (нарушением связывания вируса нейронами или особенностями вирусной репликации), но это говорит о высокой специфичности факторов, вовлекаемых во взаимодействие вируса и клетки. Одной из универсальных мишеней для вирусов являются клеточные тирозин-протеинкиназы. Благодаря сопряжению с ключевыми звеньями метаболизма их экранирование вирусными белками не только подавляет важнейшие функции клеток, но и возбуждает реакции, обслуживающие вирусную репликацию.
Апоптоз. В отличие от цитолиза, который всегда пассивен, возникая при грубых повреждениях плазматической мембраны, апоптоз является активным, генетически запрограммированным отмиранием клеток. Есть факты, позволяющие думать, что изменение соотношений между ростовыми и апоптозными потенциями клеток влияет на цитопатический эффект вирусов. Гиперэкспрессия апоптозных генов усиливает гибель, тогда как блокада апоптозной программы стимулирует рост и размножение, вызывая гиперпролиферацию (онкогенную трансформацию) клеток.
Известно более десятка вирусных генов, которые кодируют факторы, усиливающие апоптозный процесс. Обнаружены они и у вирусов человека (аденовирусы, герпесвирусы, папилломавирусы, вирусы гриппа, ВИЧ-вирусы, вирус гепатита В и др.). В ряде случаев апоптоз коррелирует с усилением экспрессии р53 — гена, претендующего на одну из ключевых позиций в индукции апоптоза. Гибель клеток удается предотвратить при помощи продукта протоонкогена bcl-2, усиливающего клеточную пролиферацию. Предложена гипотеза, согласно которой гибель CD4 Т-лимфоцитов при ВИЧ-инфекции связана с апоптозом. Пусковым фактором служит взаимодействие вирусного суперкапсидного гликопротеина gp120 с CD4-рецепторами лимфоцитов — сигнал, запускающий апоптозный механизм подобно рецепторзависимому апоптозу в системах Fas-FasL и ТНФ-ТНФR. Пока это единственный пример такого рода; в остальных случаях для индукции апоптоза требуется проникновение вируса в клетку и хотя бы частичная экспрессия его генома.
Опережающая смерть клетки, несущей зачаток вируса — исход, выгодный скорее хозяину, чем вирусу. Неслучайно об этом говорят, как об альтруистическом самоубийстве, подразумевая, что клетка предпочитает погибнуть, исключив возможность размножения потенциально опасного вируса. Логика подсказывает, что вирусы должны обладать и альтернативным механизмом, который, продлевая жизнь клеток, создавал бы условия для воссоздания как можно более многочисленного вирусного потомства в литическом цикле или обеспечивал переход вируса в латентное состояние (персистенцию). Действительно, некоторые вирусы способны блокировать апоптозный ответ на собственную инфекцию. Это достигается разными способами, но их принцип сводится к двум механизмам — повышение экспрессии антиапоптозных (ростстимулирующих) генов клетки и инактивация эффекторных молекул апоптоза. К первой группе можно отнести стимуляцию протоонкогенов типа bcl-2 (это, в частности, наблюдается при персистенции вируса Эпстайна—Барр в В-лимфоцитах), ко второй — блокаду Р53, продукта апоптозного гена р53 (Е6 протеин вируса папилломы человека — см. «Папилломавирусы»).
Образование симпласта (синцития). Феноменология сводится к слиянию нескольких клеток с образованием поликариона — многоядерной нежизнеспособной клетки. Образование симпласта характерно для сложных (оболочечных) вирусов и обусловлено рецепторзависимой цитотропностью суперкапсидных белков. Симпласт образуется двумя способами — внешним и внутренним (рис. 3).

Слияние снаружи описано для парамиксовирусов (возбудители кори, парагриппа, паротита, респираторно-синцитиальный вирус) и наблюдается при множественном заражении благодаря одновременному взаимодействию вируса (вириона) с несколькими клетками. Это вызывает их агрегацию, а затем слияние клеточных мембран. И то и другое не требует синтеза вирусных белков, проявляясь в первые часы после заражения клеточных культур. Слияние изнутри — более универсальный процесс, известный для многих оболочечных вирусов. В этом случае образованию симпласта предшествует синтез и включение вирусных белков в клеточные мембраны. Такие белки рецептируются соседними клетками, индуцируя их агрегацию. Феномен возникает при низкой дозировке инфекта и выявляется на поздних этапах репликации.
Опосредованная цитопатогенность. Гибель клеток связана не только с прямым воздействием вируса. В этом могут быть повинны и эффекторы иммунитета, атакующие зараженные клетки (см. «Механизмы противоинфекционного иммунитета»). Сложные (оболочечные) вирусы образуют суперкапсид из клеточных мембран, заблаговременно встраивая в них собственные компоненты. Но экспрессируются и антигены, представляющие глубинные элементы вириона, и даже функциональные белки, которые не входят в состав вирусных частиц. Они выныривают на поверхность клеток в комплексе с молекулами главного комплекса гистосовместимости — HLA. Клетки, экспрессирующие вирусные эпитопы, несут на себе отпечаток чужеродности, превращаясь в мишень для антител и Т-лимфоцитов. Фиксируясь на зараженных клетках, антитела подключают к цитолизу комплемент и клетки-эффекторы, располагающие рецепторами к иммуноглобулинам (макрофаги, естественные киллеры, нейтрофилы). Это хорошо моделируется in vitro, но действительное отношение антителозависимых реакций к поражению вирусинфицированных клеток остается неясным. Впрочем, антитела реально претендуют на участие в патогенезе вирусных инфекций благодаря другому механизму — иммунокомплексному воспалению. При гиперпродукции вирионов (антигенов) и антител образуется избыток циркулирующих иммунных агрегатов, которые оседают на базальных мембранах микрососудов и, активируя комплемент, вызывают острое воспаление. Особенно опасно поражение почек — гломерулонефрит, который иногда осложняет вирусные инфекции.
В этом отношении поучительна история с вирусом лимфоцитарного хориоменингита (ЛХМ). Его главный природный резервуар — мелкие грызуны, а самый распространенный источник инфекции для человека — мыши. ЛХМ взрослых мышей не отличается от ЛХМ человека, протекая как острая инфекция с преимущественным поражением ЦНС. Иной результат дает заражение новорожденных мышей. В большинстве они не только не погибают, но остаются внешне здоровыми, поддерживая персистенцию вируса. Лишь позже (не ранее 6-месячного возраста) у части мышей появляются признаки заболевания, но совсем иного, чем у остроинфицированных взрослых животных. В этом случае доминируют симптомы хронического иммунокомплексного поражения почечных клубочков — смертельного гломерулонефрита. Дело в том, что новорожденный организм иммунологически толерантен к вирусу, позволяя ему беспрепятственно размножаться. Вирус ЛХМ лишен прямой цитотоксичности, и его репликация сама по себе не ведет к патологии. Но образование антител, которое постепенно набирает силу, ведет к появлению иммунных комплексов, запускающих патологический процесс. Мыши, которые после рождения сохраняют ареактивность к вирусу, превращаются в пожизненный резервуар инфекции.
Изучение ЛХМ преподнесло еще один сюрприз. Если взрослых мышей заразить в мозг высокой дозой вируса ЛХМ, а через несколько дней ввести иммунодепрессант (например, циклофосфамид), большинство из них выживает, хотя размножение вируса продолжается. Напротив, если новорожденным мышам-вирусоносителям инокулировать в брюшную полость лимфоциты мышей, иммунных к ЛХМ, они быстро погибают от острого менингита в результате Т-опосредованного цитопатического эффекта. Т-лимфоциты вызывают гибель зараженных клеток, индуцируя цитолиз или апоптоз. При активации они секретируют цитокины (гамма-интерферон и др.), которые привлекают и стимулируют макрофаги, побуждая их к разрушительным действиям.
Все это необходимо для реализации противовирусной резистентности (подробнее об этом см. «Механизмы противоинфекционного иммунитета»). Здесь мы лишь вновь обращаем внимание на диалектику подобных реакций. При гиперболизации (особенно если вирус обладает слабой вирулентностью) они обретают болезнетворность, поддерживая цитопатический процесс. Значение этого механизма реально при вирусных инфекциях, патогенез которых не удается связать с прямой цитотоксичностью возбудителя, как это было продемонстрировано на модели вируса ЛХМ.
Трансформация. Под трансформацией понимается появление клеток с опухолевыми (онкогенными) потенциями. Такие клетки обретают бессмертие (возможность неограниченно долго перевиваться in vitro) и склонность к бесконтрольной пролиферации. В классическом варианте трансформация обусловлена активацией генов пролиферации (онкогенов, или протоонкогенов) — нормального атрибута генетического аппарата всех клеток. Некоторые вирусы несут собственные онкогены, хотя в анамнезе многие из них тоже имеют клеточное происхождение.
Большинство трансформирующих вирусов человека и животных относятся к ДНК-вирусам, обладая способностью встраивать свои гены в хромосомы инфицированной клетки. Это характерно для непермиссивных клеток, которые благодаря этому избегают деструктивного действия вирусов. Другим вариантом является заражение дефектными вирионами (см. ниже). В таких условиях экспрессируется ограниченное число вирусных генов, в том числе таких, продукты которых нарушают регуляцию клеточного цикла, инактивируя один или несколько супрессорных белков (например, Rb, p53). Именно в этот момент, т.е. в период ослабления митотического процесса, наиболее вероятны спонтанные и индуцированные мутации.
В действительности такие действия предназначены не для туморогенеза, а для вирусной репродукции. Дело в том, что большинство ДНК-вирусов используют для репликации ДНК механизмы клетки-хозяина. Это рационально, но проблема в том, что клеточные факторы, задействованные в данном процессе, экспрессируются обычно во время синтеза собственной ДНК (S-фаза клеточного цикла).
Вирусам невыгодно дожидаться начала митоза, тем более что многие клетки-мишени дифференцированы и не делятся (например, поверхностные эпителиальные клетки). Чтобы преодолеть это, ДНК-вирусы научились продуцировать факторы, побуждающие к вхождению зараженных клеток в митотический цикл. К ним, в частности, относятся ранние (т.е. синтезируемые первыми) вирусные белки, блокирующие ингибиторы митоза. Например, аденовирусный белок Е1b (так же, как белки папилломавирусов и герпесвирусов) связывается с клеточным белком р53, который препятствует началу S-фазы. Это выводит клетку из-под негативного контроля, позволяя экспрессировать факторы, необходимые для репликации. По аналогии другие вирусные белки связывают клеточный белок pRB, высвобождая фактор транскрипции, усиливающий образование белков, необходимых для деления клеток.
Нарушая нормальную регуляцию митотического цикла, такие вирусы обладают онкогенным потенциалом. Это особенно характерно при интеграции вирусного генома в клеточную ДНК, когда транскрипции подвергаются прежде всего ранние гены вируса. Действительно, два упомянутых клеточных фактора (р53 и pRB) известны как супрессоры опухолевого роста, а канцерогенными (для человека) ДНК-вирусами являются папилломавирусы, герпесвирусы (вирус Эпстайна—Барр) и гепаднавирусы (вирус гепатита В). Впрочем, об этом принято говорить более осторожно: их эффект ассоциирован с развитием строго определенных форм рака.
Из РНК-вирусов лишь ретровирусы вызывают феномен трансформации, являясь причиной ряда злокачественных опухолей животных и одной из редких форм Т-клеточного лейкоза человека (вирусы HTLV-1 и HTLV-2). Впрочем, такое исключение скорее подтверждает общее правило: для репликации ретровирусы используют смешанную ДНК-РНК-стратегию, внедряя в хромосомы свой ДНК-прогеном, который образуется путем обратной транскрипции вирионной РНК. Интеграция с вирусными генами может дестабилизировать геном клетки, возбуждая онкогены. Есть подозрение, что к трансформации предрасполагает и вирусиндуцированное ослабление апоптозного процесса, создающее дисбаланс между пролиферацией и гибелью клеток. Парадоксально, но именно вирусо-генетическая теория рака привела к обобщающей концепции об эндогенных онкогенах, продукты которых, являясь физиологическими стимулами пролиферации, способны выходить из-под негативного контроля, возбуждая избыточный рост клеток. Вирусы вписываются сюда как один из возможных коиндукторов клеточных онкогенов, а также как фактор, нарушающий равновесие между пролиферацией и апоптозом.
Персистенция
Клинический облик многих вирусных инфекций связан с персистенцией — длительным присутствием возбудителя в организме. В основе персистенции лежит способность вирусов закрепляться в клеточных популяциях, используя для этого разнообразные приемы — от «замораживания» собственных генов в хромосомах клетки-хозяина (вирогения) до агрессивного вмешательства в систему индукторов и эффекторов иммунитета.
Напомним, что развитие (онтогенез) вирусов включает две фазы — внеклеточную и внутриклеточную. Вне клеток вирус представлен структурно зрелой частицей — вирионом. Попадая в чувствительные клетки, вирион распадается (дизъюнкция), высвобождая нуклеиновую кислоту, которая заставляет клетку реплицировать вирусные субкомпоненты, необходимые для сборки новых вирионных частиц. Такой цикл, нередко ведущий к гибели клеток, называется продуктивной, или репликативной, инфекцией. Именно этот процесс имел в виду М. Бернэт, рассуждая о фатальных исходах острых вирусных заболеваний. Он говорил о них как об экологической катастрофе не только для хозяина, но и для вируса. Действительно, чтобы существовать, вирусу выгодно избегать или хотя бы сглаживать конфликты с хозяином. Следуя логике, для этого он должен найти относительно устойчивый организм, с которым мог бы вступить в симбиоз в виде продуктивной инфекции или вирогении.
Имеется множество наблюдений, доказывающих способность вирусов длительно сохраняться в клетках in vitro и in vivo. У человека персистируют герпесвирусы, вирусы гепатитов В и С, кори, полиомиелита, ретровирусы и др. Из клеточных культур от бессимптомных носителей были случайно выделены не известные ранее вирусы человека и животных — аденовирусы, риновирусы и пр. Открытие ряда опасных вирусов явилось результатом случайных внутрилабораторных заражений при работе с культурами клеток здоровых животных, прежде всего обезьян. Именно так были обнаружены вирусы геморрагических лихорадок Ласса и Марбург. Вирус бешенства сохраняется в летучих мышах, выделяется от здоровых собак и песцов. Арбовирусы персистируют в кровососущих членистоногих и, попадая к человеку, вызывают тяжелейшие заболевания — энцефалиты и геморрагические лихорадки. До сих пор не исключена вероятность «внечеловеческих» резервуаров гриппозного вируса, где, подвергаясь генетическим перестройкам, он готовит себя к очередным эпидемиям.
Высокочувствительные методы иммунохимического и молекулярно-генетического анализов значительно расширили возможности изучения персистентных инфекций. Если вирусологи прошлого теряли вирус тотчас после его проникновения в клетку и не видели вплоть до образования новых вирионов, то сегодня за вирусом ведется непрерывное наблюдение, так как зараженные клетки всегда содержат вирусные гены и белки. Словом, как бы ни старался спрятаться вирус, он, подобно герою-невидимке Г. Уэлса, обязательно оставляет следы, пусть незначительные, но уловимые современными методами.
Лет тридцать назад французский вирусолог О’Дюруа говорил о персистенции как о джентльменском соглашении между вирусом и клеткой, подписи которого известны, но условий которого мы не знаем. Сегодня можно говорить и об условиях, т.е. о механизмах вирусного симбиоза.
Персистенция на уровне изолированных клеток
Персистенция вирусов имеет глубокие эволюционные корни. Ее классической моделью является фаговая лизогения, по сути симбиоз между бактериями и их вирусами. Большинство лизогенных (умеренных) фагов длительно сохраняются в бактериях, внедряя свои гены в хромосому и подавляя их транскрипцию при помощи репрессорного белка, закодированного в собственном (фаговом) геноме. Инактивация гена-репрессора снимает блокаду, индуцируя репликативную инфекцию.
Аналогичный механизм (интегративная вирогения) для вирусов человека и животных был постулирован Л.А. Зильбером в рамках вирусо-генетической теории рака. Действительно, подобно лизогенным фагам, они могут длительно персистировать в составе клеточных хромосом в полностью или частично законсервированном виде. В этом им помогают не только собственные репрессоры, но и зараженные клетки, которые продуцируют факторы, контролирующие транскрипцию вирусных генов. Для некоторых вирусов (ретровирусы, гепаднавирусы) интегративная вирогения является обязательным условием продуктивной инфекции. На этой основе возникло даже представление об интеграционных болезнях, признающее патогенетическую значимость интегративного процесса, т.е. включения вирусных генов в клеточные хромосомы.
Но вирогения возможна и без интеграции. Это характерно для РНК-вирусов, лишенных механизма обратной транскрипции (переписывания генетической информации с РНК на ДНК), т.е. в принципе не способных к объединению с ДНК. Так ведет себя, например, вирус гепатита С. Вирус кори (представитель семейства парамиксовирусов) сохраняется после острой инфекции в центральной нервной системе. Подавление его репродукции объясняется тем, что в нейронах не синтезируется М-белок, необходимый для сборки вирионов. Это ведет к медленному накоплению вирусных субкомпонентов (нуклеокапсидов) и развитию подострого склерозирующего энцефалита. Впрочем, и ДНК-вирусы способны персистировать автономно от хромосом, подобно бактериальным плазмидам. Это, в частности, характерно для герпетической инфекции нейронов (см. «Герпесвирусы»).
Другой механизм уклонения от острого конфликта связан с дефектными вирусами. Возникая в результате мутационных делеций, некоторые из них (так называемые дефектные интерферирующие частицы — ДИ-частицы) не только сохраняют способность к самовоспроизведению, но реплицируются быстрее полноценных вирусов, используя их в роли помощников. Накопление ДИ-частиц препятствует избыточному размножению вирулентных вирусных клонов, предрасполагая к персистенции: цитопатический вирус и ДИ-частицы размножаются, регулируя друг друга. Многочисленными исследованиями разных вирусов показано, что ДИ-частицы могут накапливаться в организме зараженных животных, защищая от вирулентного вируса. Последний не элиминируется, а переходит в латентное состояние, иногда полностью прекращая репликацию. В связи с тем, что в различных клеточных системах скорость накопления ДИ-частиц неодинакова, степень загрязнения вирусных препаратов ДИ-частицами сильно варьирует. Например, для получения определяемого количества ДИ-частиц пикорнавирусов требуются десятки последовательных пассажей, тогда как вирус бешенства продуцирует большое количество ДИ-частиц уже при первых посевах. С этим, в частности, связан парадокс, отмеченный Л. Пастером при разработке антирабической вакцины: эффективность воспроизведения бешенства снижается при заражении животных большими дозами инфицированного материала (мозговой эмульсии от больных животных), когда повышается вероятность засорения вирулентного вируса ДИ-частицами.
Разновидностью дефектных вирусов являются так называемые температурозависимые мутанты, которые не реплицируются при 37оС (т.е. нормальной температуре тела), но размножаются при повышенных температурах. Альтернативой являются риновирусы — главные возбудители малых простудных заболеваний (острого катара верхних дыхательных путей). Они тоже температурозависимы, но предпочитают пониженную температуру. Персистируя в назальном эпителии, где и без того «холодно», риновирусы активируются при дополнительном охлаждении (простуда!).
Проблема вирусной персистенции получила отражение и в концепции апоптоза. Подразумевается, что при так называемом альтруистическом самоубийстве, реагируя на вирусную инвазию, клетки предпочитают погибнуть, чтобы опередить репликацию вируса, опасного для своих соседей и организма в целом. Некоторые вирусы блокируют клеточный апоптоз, создавая условия для продуктивной инфекции или персистенции. Они добиваются этого, повышая экспрессию ростстимулирующих генов (протоонкогенов) либо инактивируя эффекторы апоптоза.
Персистенция на уровне организма
Чтобы закрепиться в организме, вирусу недостаточно «договориться» с клеткой. Как чужеродный (антигенный) объект он подвергается иммунной атаке, нацеленной на уничтожение вируса и зараженных им клеток. Это означает, что на уровне организма персистенция предполагает еще одно условие: вирус должен избежать элиминирующих реакций в системе иммунологического надзора. Главное здесь — выживание не свободного вируса, а зараженных клеток, т.е. персистенция не самого вируса, а содержащих его клеток.
Персистенция затрагивает практически все факторы антивирусной защиты. Многие обобщения гипотетичны и служат скорее ориентиром для дальнейших исследований, которые могут утвердить их или опровергнуть. Но они представляют несомненный интерес, отражая стремление расшифровать одно из базисных понятий вирусологии. Мы сконцентрируем внимание на взаимоотношениях вирусов с Т-лимфоцитами, так как именно от них больше всего зависит судьба инфицированных клеток. Антитела предупреждают заражение клеток, но не спасают от внутриклеточного вируса. Они необходимы для успешного завершения острого конфликта, но не эффективны против утвердившейся инфекции.
О противовирусном иммунитете говорится в специальном очерке (см. «Механизмы противоинфекционного иммунитета»). Здесь мы лишь напомним о том, что необходимо знать для обсуждения антииммунитетной стратегии вирусов (рис. 4). Ключевым является представление вирусных антигенов Т-лимфоцитам в комбинации с молекулами главного комплекса гистосовместимости (у человека — HLA) на поверхности зараженных клеток или клеток, поглотивших вирусные антигены. На этом этапе происходит селекция антигенчувствительных клонов, которые, подвергаясь дифференцировке и пролиферации, превращаются в Т-эффекторы, настроенные (коммитированные) против HLA-презентируемых эпитопов.

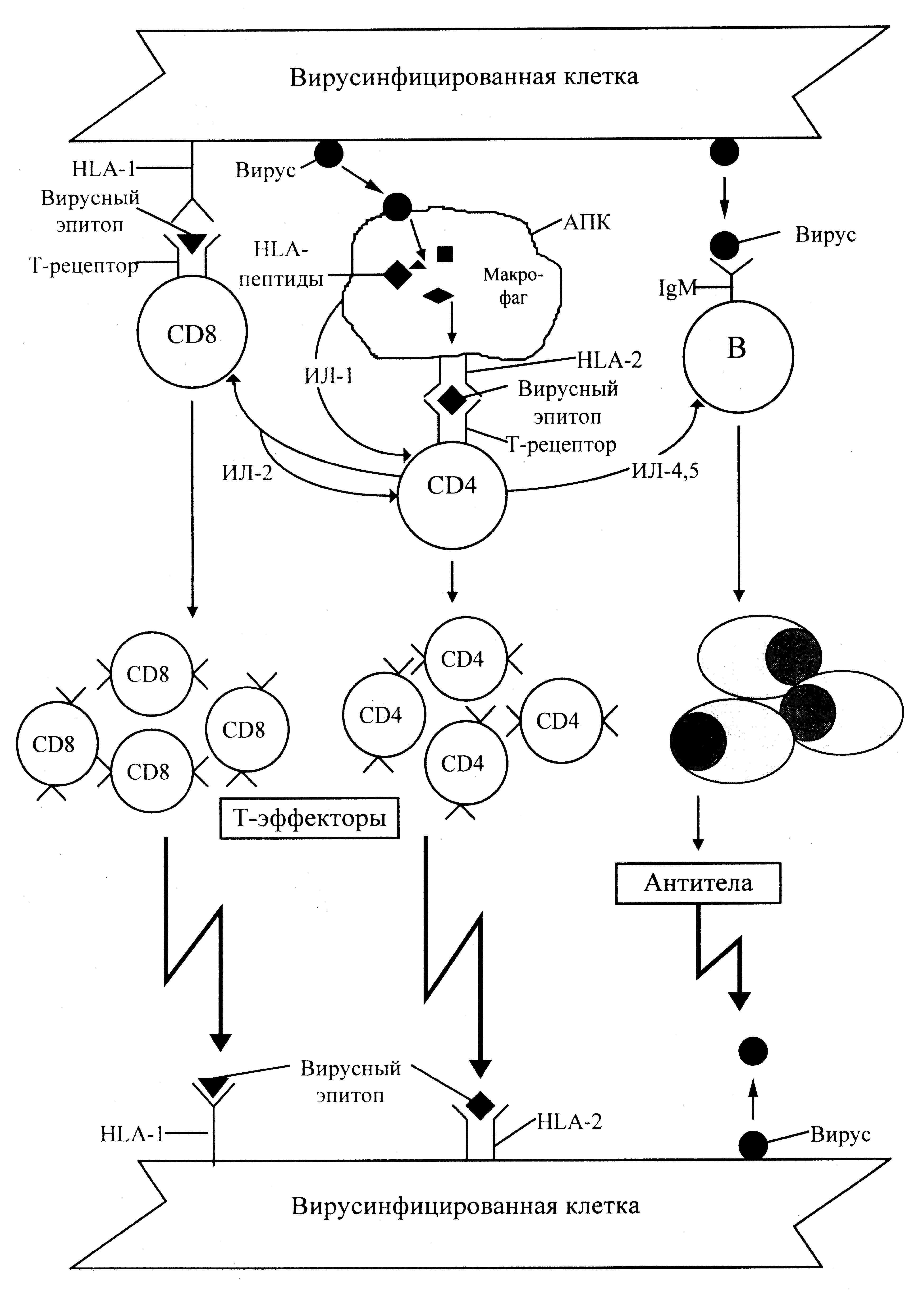
Для созревания Т-эффекторов необходимы дифференцировочные и ростстимулирующие сигналы хелперных CD4 Т-лимфоцитов. Но перед этим T-хелперы сами подвергаются активации, превращаясь в источник разнообразных цитокинов. Главная роль принадлежит Тh1. Они помогают активации цитотоксических Т-лимфоцитов и других эффекторных клеток, прежде всего макрофагов.
Реализация иммунного ответа против внутриклеточных вирусов связана с цитотоксическими Т-лимфоцитами (СТL). Неспецифические клетки-эффекторы дополняют их антивирусную активность.
Из сказанного вытекают два правила, следуя которым вирус может избежать уничтожения. Во-первых, этому способствует слабый иммунный ответ, недостаточный для элиминации вируса и вирусинфицированных клеток. Во-вторых, вирус может пассивно или активно уклоняться от взаимодействия с эффекторами иммунитета, прежде всего с цитотоксическими Т-лимфоцитами. Иными словами, антииммунитетная стратегия вирусов может иметь профилактическую направленность (подавление индукции иммунного ответа) или проявляется после того, как хозяин уже накопил определенный запас антивирусных средств (подавление реализации иммунного ответа).
Подавление индукции иммунного ответа. Идеальный способ для исключения иммунного ответа — полная блокада вирусных генов, персистирующих внутри клетки: молчащие гены не вызывают синтеза белков, и, следовательно, клетка не будет мечена вирусными антигенами. Но в таком случае вирус лишился бы экологического пресса, перестав эволюционировать. Отсюда любой вирус, претендующий на закрепление в природе, обязан полностью или частично реплицироваться, а персистенция — лишь хорошее подспорье для того, чтобы рано или поздно проявить свой репликативный потенциал.
Одним из примеров стабилизации вирусного инфекта на фоне ослабления иммунной реакции является внутриутробное инфицирование, создающее прецедент для негативной селекции (элиминации) незрелых Т-лимфоцитов, коммитированных против вируса. Это вызывает толерантность, т.е. иммунологическую ареактивность против вируса. Такая возможность обсуждается в связи с трансплацентарным заражением вирусом гепатита В (HBV), которое в 90% случаев ведет к хроническому вирусоносительству; у взрослых острый гепатит В завершается персистенцией вируса лишь в 10% случаев. Врожденная HBV-инфекция сочетается с ослаблением ответа на нуклеокапсидные HBV-эпитопы; cпособность к образованию антител против поверхностного антигена (HBsAg) сохраняется (см. «Вирусы гепатита»).
Относительная толерантность к вирусным антигенам возникает и у взрослых. Замечено, например, что при хроническом гепатите В количество анти-HBV CTL гораздо меньше, чем у больных острым гепатитом, а их противоантигенный (точнее, противоэпитопный спектр) значительно сокращен. Причиной может быть истощение антивирусных Т-клонов избытком вирусных антигенов или разрушительная аутоагрессия против СТL с фиксированными на их поверхности вирусными пептидами (результат связывания свободных HBV-пептидов молекулами HLA-1).
Другим механизмом может быть недостаточная презентация вирусных антигенов в системе главного комплекса гистосовместимости. Обладая высоким полиморфизмом, молекулы HLA отличаются по сродству к различным пептидам и способности представлять их Т-лимфоцитам. Это означает, что индивиды с разным HLA-фенотипом могут неодинаково реагировать на один и тот же вирус. В реальных ситуациях, когда в реакцию втягивается множество антигенных эпитопов и Т-клонов, влияние подобного фактора на иммунный ответ маловероятно, но это может иметь значение, если речь идет о реактивности к наиболее важным (протективным) антигенам. Значимость такого механизма возрастает и при слабом иммунном ответе, для которого характерна олигоклональность, т.е. вовлечение в реакцию ограниченного числа лимфоцитарных клонов на лимитированный спектр эпитопов. Это довольно типично для хронических вирусных инфекций.
Но вирусы способны и активно блокировать презентацию собственных антигенов. Они добиваются этого, снижая экспрессию HLA и других иммуновспомогательных молекул на поверхности зараженных клеток. По примерным подсчетам, для индукции Т-ответа требуется 200 HLA-пептидов; за этим порогом клетки незаметны для Т-лимфоцитов. Способность к сокращению числа молекул HLA обнаружена у аденовирусов, цитомегаловируса, вируса Эпстайна—Барр. Эффект может быть непрямым. В этом случае он опосредован через подавление секреции или блокаду цитокинов, стимулирующих экспрессию антигенпредставляющих молекул, а также через ослабление чувствительности вирусинфицированных клеток к такого рода медиаторам. Это установлено для вируса гепатита В: его кор-антиген (HBcAg) снижает синтез альфа-интерферона, а ДНК-полимераза подавляет клеточные реакции на альфа- и гамма-интерфероны. То же самое известно для нуклеокапсидного белка вируса гепатита С, который вызывает значительное подавление альфа-интерферона у больных хроническим гепатитом С.
Наконец, вирусы обладают способностью снижать активность Т-лимфоцитов. Существует масса экспериментальных и клинических данных, полученных для разных вирусов (герпесвирусы, аденовирусы, орто- и парамиксовирусы и др.). Они носят фактологический характер и лишь с большой натяжкой могут быть экстраполированы на персистенцию. Примером патогенетически значимой иммунодепрессии служит антихелперная (анти-CD4) активность вируса иммунодефицита человека. Но и этот механизм, срабатывающий с большим опозданием (когда вирус уже закрепился в организме), вряд ли может быть главной причиной стабилизации ВИЧ-инфекции, которой на первых порах сопутствует выраженный иммунный ответ (см. «Вирус иммунодефицита человека»).
Подавление реализации иммунного ответа. За редким исключением вирусам не удается избежать иммунной реакции. Поэтому они вынуждены персистировать на фоне более или менее значительного накопления эффекторов иммунитета — антител и Т-лимфоцитов. Если антитела порой даже способствуют вирусоносительству, сдерживая чрезмерную агрессивность свободных вирусов, то клетки, меченные вирусными антигенами, служат отличной мишенью для СТL и, казалось бы, подлежат обязательному уничтожению. Тем не менее очищение организма от вирусов часто запаздывает, а в ряде случаев остается незавершенным. В идеале вирус должен стать незаметным для антител и Т-эффекторов, а реально — избежать с ними острого конфликта, обрывающего инфекцию. Вирусы добиваются этого разными способами, от простых и очевидных до весьма изощренных. Впрочем, не более изощренных, чем способы, которыми пользуется хозяин, чтобы избавиться от поселившихся в его организме инфекционных агентов.
Во-первых, вирусы могут персистировать, сокращая экспрессию своего генома ниже порога видимости Т-эффекторов либо инфицируя иммунологически привилегированные ткани, т.е. участки тела, труднодоступные для Т-лимфоцитов или наделенные способностью возбуждать их апоптоз при помощи Fas-Fasl-зависимого механизма.
Например, главным резервуаром латентной ВИЧ-инфекции служат макрофаги, которые в отличие от CD4 Т-лимфоцитов не поддерживают активной репликации вируса. Вирусы простого герпеса и ветряной оспы замораживают большинство своих генов в нейронах регионарных ганглиев чувствительных нервов. Цитомегаловирус персистирует в макрофагах и слюнных железах, укрываясь за гематосаливарным барьером. Вирус кори прячется в нейронах головного мозга. Символично выражение: мозг как мусоросборник вирусов. Во-первых, гематоэнцефалический барьер — надежная преграда для лимфоцитов, а, во-вторых, нейроны, лишенные пролиферативных потенций, малопригодны для вирусной репликации. Альтернативой являются гепатоциты, которые, минуя микроваскулярные (гематотканевые) барьеры, напрямую контактируют с кровью и легко доступны для Т-лимфоцитов. Отсюда понятно, почему персистирующие гепатотропные вирусы (HBV, HCV) прячутся не только в гепатоцитах, но и в других клетках, включая иммунологически привилегированные зоны. Возможно, что, выходя из укрытия, они реинфицируют гепатоциты и одновременно активируют Т-клетки памяти, тонизируя патогенетический механизм хронического гепатита.
Парадоксально, но фактором, способствующим персистенции, могут быть интерфероны, исходно нацеленные на уничтожение внутриклеточного вируса. Это связано с отбором вирусных мутантов, резистентных к интерфероновой блокаде вирусной репликации. Повышенной устойчивостью к интерферонам обладают мутанты по пре-С-гену вируса гепатита В. Больные, инфицированные такими штаммами, хотя и отвечают на интерферонотерапию, но чаще дают обострения. Не одинаковы по чувствительности к интерферону и генотипические разновидности вируса гепатита С. Это также влияет на эффективность лечения его хронических форм.
Устойчивость к интерферонам — не единственный пример иммунорезистентности вирусов. Уклонению от антител и Т-лимфоцитов способствует изменчивость вирусных антигенов, которая ведет к образованию так называемых ускользающих мутантов (англ. escape mutants). Это связано с высокой скоростью точечных мутаций по протективным эпитопам и HLA-пептидам, представляющим антигены в системе Т-клеточного иммунитета. Появление антигенных мутантов в ходе инфекционного процесса обнаружено для вирусов иммунодефицита человека, гепатита В и С, простого герпеса, полиомиелита. В таких случаях вирус представлен в организме совокупностью неоднородных клонов (квази-видов), которые подвергаются селекции под давлением иммунологического пресса.
Крайним проявлением эпитопной изменчивости служит полная потеря базисной специфичности, когда мутантный антиген не связывается с антителами и рецепторами Т-лимфоцитов против исходных (диких) вирусных клонов. Это обеспечивает выживание вируса даже в условиях жесткой иммунной атаки. У больных HBeAg-позитивным хроническим гепатитом В почти все HBV-клоны обновляются через 5—6 лет, мутируя по пре-S-гену и меняя соответствующие эпитопы. Это позволяет вирусу персистировать после элиминации дикого штамма. Известны наблюдения, когда поверхностный антиген (HBsAg) вируса гепатита В циркулировал в крови, но не улавливался стандартными анти-HBs-антителами из-за мутаций всего по 1—2 аминокислотам. Такие случаи квалифицировались как ни А—В гепатит, и лишь по генетическим маркерам ставился правильный диагноз. Нейтрализующая активность антител, полученных на ранней стадии хронического гепатита С, прогрессивно падает по отношению к вирусным штаммам, изолированным в более поздние сроки. Чем меньше разрыв между получением вируса и антител от одного и того же больного, тем сильнее их антивирусный эффект. Мутанты, ускользающие от эффекторов иммунитета, чрезвычайно характерны для ВИЧ-инфекции. Скорость мутирования ВИЧ (особенно по главному протективному антигену, gp120) необыкновенно высока, обеспечивая широкие возможности для селекции иммунорезистентных вирусных клонов.
Интригующую разновидность Т-эпитопных модификаций представляют вирусные мутанты-антагонисты. Сохраняя способность связываться с Т-клеточными рецепторами, они вместо активации вызывают частичную или полную анергию CTL. Ее отношение к вирусной персистенции становится реальным, если учесть, что такая анергия может быть длительной, а зараженные клетки способны экспрессировать на поверхности дикие и мутантные вирусные эпитопы. Последние, интерферируя с СТL, защищают не только свой, но и родительский (немутантный) вирус. Т-антагонизм (он обнаружен у вирусов гепатита В и иммунодефицита человека) не связан с физической блокадой Т-рецепторов, так как для его воспроизведения требуется ничтожная дозировка мутантных Т-эпитопов, которая явно недостаточна для экранирования антигенных рецепторов. Следует скорее думать о фармакологическом эффекте, т.е. о блокаде образования медиаторов, транслирующих сигналы с Т-рецепторов.
Еще один механизм антииммунитетной вирусной стратегии — нейтрализация цитокинов и их рецепторов. К числу цитокинов относятся важнейшие регуляторы индукции и реализации иммунного ответа, большинство из которых продуцируется активированными макрофагами и Т-лимфоцитами. Отсюда вряд ли случайно, что некоторые вирусы (прежде всего ДНК-вирусы с крупным геномом) научились дискриминировать этот механизм, перехватывая цитокины структурными аналогами их рецепторов (вироцепторы) или экранируя цитокиновые рецепторы при помощи пептидов, сходных с их естественными лигандами — цитокинами (вирокины). Так, поксвирусы кодируют пептиды, гомологичные рецепторам для туморонекротического фактора, интерлейкина-1, гамма-интерферона, альфа-/бета-интерферонов. Они (вироцепторы) выделяются из инфицированных клеток и, связывая цитокины, опережают их эффекты в системе антивирусной защиты. Герпесвирусы кодируют структурные аналоги рецепторов хемокинов — мощных хемоаттрактантов для клеток-эффекторов воспаления и иммунитета. Хемокиновые вироцепторы экспрессируются на поверхности зараженных клеток и, отвлекая на себя часть хемокинов, снижают интенсивность антивирусной реакции. Аденовирусы синтезируют белки, интерферирующие с цитотоксической активностью туморонекротических факторов CТL, естественных киллеров и макрофагов. Фактор, сходный с интерлейкином-1, обнаружен у вируса Эпстайна—Барр; одна из его функций связана с ослаблением реакций в системе Т-клеточного иммунитета.
Резюмируя, можно сказать, что благодаря необыкновенно плотной коэволюции вирусов и их хозяев возникли удивительно изящные формы симбиотических взаимоотношений, которые прослеживаются на всех уровнях организации вирусной материи — от бактериофагов до вирусов высших животных. Персистенция сочетает в себе элементы пассивной и активной самозащиты. Они неодинаково используются разными вирусами, имеют различную адаптивную силу и неравноценные патогенетические последствия.
Активация персистентных вирусов
Вирусная латенция динамична. Равновесие между вирусом и клеткой, вирусом и организмом может сдвигаться в пользу хозяина или паразита, создавая для каждого из них более или менее серьезные проблемы. По этому поводу А.Львов (французский генетик и вирусолог русского происхождения) остроумно заметил, что взаимоотношения между умеренными (т.е. персистентными) фагами и бактериями подобны брачному союзу: он кажется прочным, но под давлением обстоятельств рушится, и бывшие друзья становятся врагами. Нечто подобное справедливо и для медицинских вирусов. Подвергаясь активации, они реализуют свою болезнетворность, внося существенный вклад в инфекционную патологию.
Причины активации латентных вирусов (так же, как корни самой персистенции) следует анализировать по двум направлениям — на уровне клеток и на уровне организма. И в том и другом случае могут быть виновны вирус или хозяин, но в основе всегда лежит ослабление контроля над вирусом. Это переводит латенцию на рельсы продуктивной инфекции или хотя бы дерепрессирует вирусные гены, продукты которых приносят прямой или опосредованный вред хозяину. Вирусы меняются в результате мутаций, генетических рекомбинаций, генотипического или фенотипического смешивания с вирусами-помощниками. Отсюда толчком к активации может быть воздействие физических и химических факторов, оксидантного стресса, суперинфекции. Например, температурозависимые мутанты оживают при повышении температуры, а риновирусы активируются при охлаждении. Для репликации вируса гепатита D требуется присутствие вируса гепатита В, некоторые из парвовирусов нуждаются в помощи аденовирусов или вирусов простого герпеса. Иногда латенцию удается нарушить при сокультивировании с чувствительными (пермиссивными) клетками.
Одним из общих условий для патогенетически значимой активации вирусов является ослабление резистентности хозяина. С наибольшей очевидностью это проявляется у больных с ятрогенной и ВИЧ-иммунодепрессией. Одной из серьезных проблем в таких случаях являются эндогенные рецидивы герпетических инфекций на фоне подавления Т-клеточного иммунитета. Впрочем, и нормально работающая иммунная система не только не гарантирует защиты от клинически значимой персистенции вирусов, но и участвует в реализации ее патогенетического потенциала. Это совершенно необходимо для вирусов, которые лишены собственной цитотоксичности, формально безвредны для изолированных клеток, но обретают болезнетворность, подключая эффекторы иммунитета.
Активация персистентных вирусов ведет к разнообразным клиническим последствиям, которые, как и при обычных инфекциях, отражают избирательную цитотропность и особенности взаимоотношений в системе «вирус-клетка». Памятуя об этом, их можно объединить в несколько групп (рис. 5). Рецидивы острых поражений типичны для вирусов с прямой цитотоксичностью (вирусы простого герпеса, ветряной оспы, опоясывающего лишая, цитомегаловирус). Хроническое течение имеет обычно иммунопатогенетическую основу (например, гепатит В). Опасность внутриутробного инфицирования возникает при вирусемии беременных, предрасполагающей к трансплацентарной (вертикальной) передаче возбудителя (цитомегаловирус, вирус гепатита В, ВИЧ). Онкогенность обычно связана с интегративными инфекциями, когда вирусы, дестабилизируя клеточный геном, способствуют экспрессии его онкогенов (точнее — протоонкогенов).

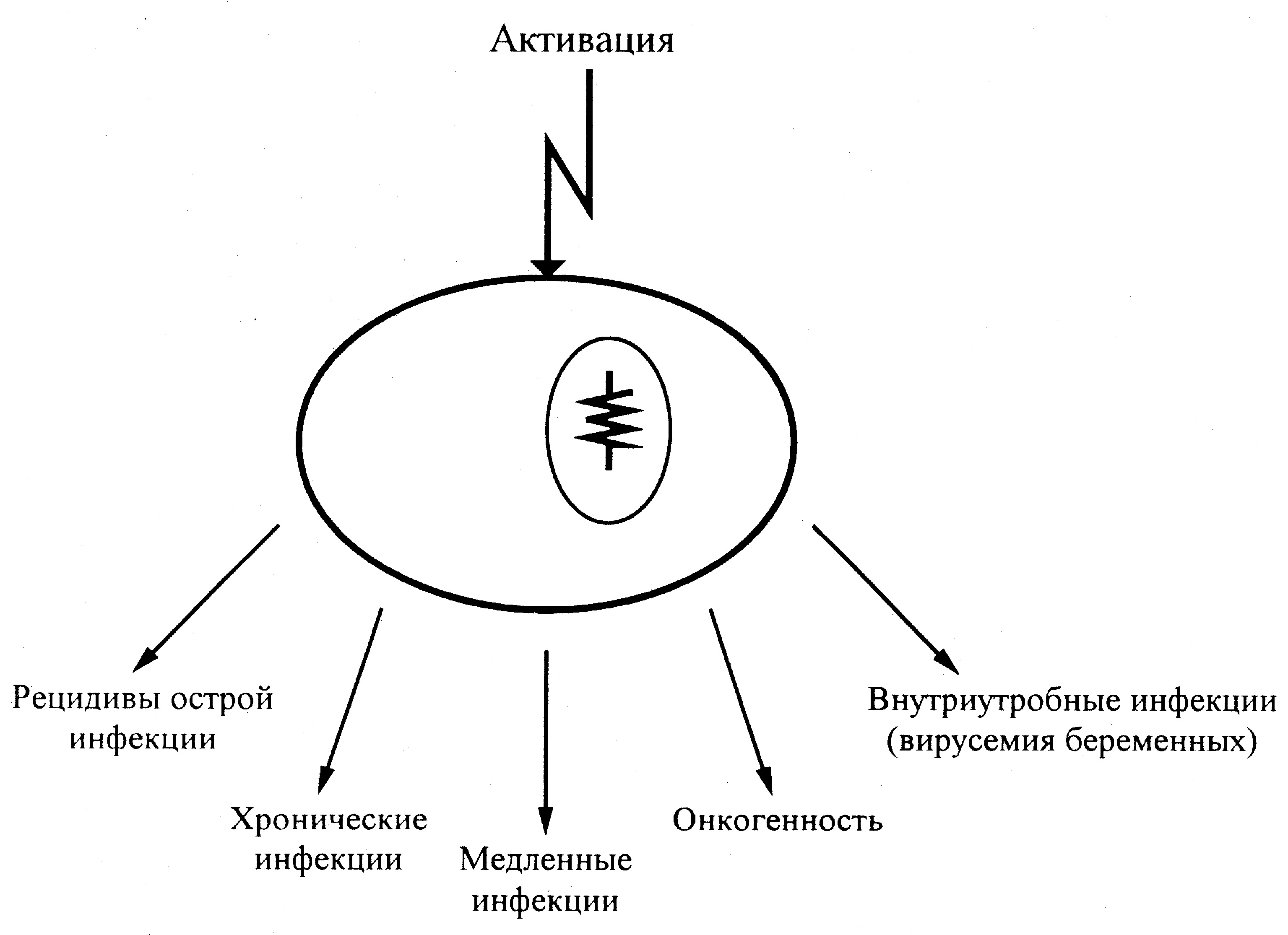
Особой категорией являются медленные нейроинфекции — подострые спонгиозные (губчатые) вирусные энцефалопатии. Они характеризуются длительным (несколько лет) инкубационным периодом и, прогрессируя без ремиссий, неизбежно ведут к смерти на фоне необратимой дегенерации нейронов. Возбудители некоторых из них совершенно уникальны: это инфекционные белки, лишенные генетического материала (ДНК или РНК). Именно поэтому они получили название прионов (сокращение двух слов — протеин и инфекционный). О прионах говорят как о неканонических вирусах, так как отсутствие нуклеиновой кислоты возводит их в ранг самостоятельной формы живой материи, заставляя вспомнить Ф. Энгельса: жизнь — есть способ существования белковых тел. Прионы устойчивы к обычным физическим и химическим воздействиям, включая ультрафиолетовое и ионизирующее облучение, ультразвук, нагревание, антисептики. Не имея нуклеиновой кислоты, они в отличие от обычных вирусов не инактивируются нуклеазами и не чувствительны к интерферону. Прионные поражения не сопровождаются воспалительной реакцией и не вызывают иммунного ответа. Поэтому они не несут специфических иммуносерологических маркеров и диагностируются лишь на основании клинических и аутопсийных признаков.
Сюрпризом явилось обнаружение прионов не только в пораженной нервной ткани (он накапливается здесь в виде амилоидных бляшек), но и в нормальных клетках, причем не только в нейронах. По-видимому, он выполняет здесь какие-то физиологические функции, о которых можно только гадать. Не исключено, например, что нормальный прион (в отличие от патогенного PrPSc-белка, он обозначается PrPC) необходим для стабилизации нейронного сообщества ЦНС, прежде всего головного мозга.
Происхождение прионовой патологии видится сегодня следующим образом. Действуя аутокаталитически, экзогенные прионы вызывают конформационные изменения в клеточном прионе, переводя его в патогенетически значимые изоформы. Обладая устойчивостью к внутриклеточным протеазам, PrPSc накапливаются в клетках, вызывая дегенеративные изменения. Полагают, что реакция носит цепной характер: молекула PrPSc cоединяется с PrPC с образованием гетеродимерного продукта, который трансформируется в две молекулы PrPSc; в следующем цикле две молекулы PrPSc связываются с PrPC, давая начало четырем PrPSc; в третьем цикле каждая из них соединяется с PrPC, генерируя восемь молекул PrPSc и т.д. Это обеспечивает экспоненциальный рост PrPSc.
Прионы являются причиной нескольких заболеваний человека (куру, болезнь Крейтцфельдта—Якоба, синдром Герстманна—Штреусслера—Шейкера, фатальная семейная инсомния), а также нескольких болезней животных (скрэпи овец и коз — отсюда акроним «Sc” в PrPSc, инфекционная энцефалопатия домашних норок, хроническое истощение одомашненных оленей и лосей, трансмиссивная губчатая энцефалопатия коров — болезнь «бешеных коров» и др.). Прионы способны преодолевать видовые барьеры, что отражает эволюционный консерватизм приона, сохранившего структурное сходство у большинства млекопитающих. Это, в частности, спровоцировало нашумевшую историю в связи с постулированной, но недоказанной возможностью заражения человека прионами болезни Крейтцфельдта—Якоба через мясо «бешеных» коров.
Избирательное поражение ЦНС заслуживает отдельного комментария. Оно не связано со специфической рецепцией прионов нейронами, так как при периферийном введении прежде чем попасть в мозг, прионы обнаруживаются в ретикулоэндотелиальной системе. Но так же как в перевиваемых клеточных линиях они никогда не накапливаются здесь в высоких титрах. Это объясняется тем, что клетки делятся чаще, чем удваивается количество прионов, а потому самостерилизуются. Размножение прионов идет очень медленно со временем удвоения примерно 5 дней, и для накопления патогенетически значимой концентрации требуются многие недели, а нередко — месяцы. Поэтому не удивительно, что прионы поражают только нейроны, которые лишены способности к делению.
Этиология медленных нейроинфекций не ограничивается прионами. Их причиной могут быть и обычные, канонические вирусы, персистирующие в ЦНС. Кстати, именно с них началось изучение этой удивительной патологии. Начало положил Б. Сигурдсон, доказавший инфекционную природу болезни висны-мэди овец, завезенных в Исландию из Германии для развития каракулеводства. Эти исследования начались в конце 1930-х гг., но лишь в 1954 г. Б.Сигурдсон сформулировал основные положения концепции о медленных инфекциях. Возбудителем висны-мэди (исл. «чахнущий» и «одышка») оказался ретравирус, чем поставил немало вопросов. Неожиданным, например, явилось то, что он отлично размножается в клеточных культурах, вызывая острый (т.е. быстрый) цитопатический эффект. Иными словами, отсроченное и медленное поражение ЦНС связано с особенностями поведения вируса в инфицированном организме, а не с его биологической уникальностью. Было установлено, что возбудитель бессимптомно персистирует в макрофагах, а его распространение сдерживается антителами. В вирусной популяции вызревают ускользающие мутанты, и не исключено, что именно они обеспечивают эволюцию процесса.
Человек не заражается вирусом висны-мэди, но у него есть свои медленные нейротропные вирусы. Прогрессирующая дегенерация нейронов головного мозга наблюдается при ВИЧ-инфекции, являясь одним из центральных проявлений синдрома приобретенного иммунодефицита. Вирус кори, персистируя в нейронах головного мозга, вызывает подострый склерозирующий энцефалит. Опасны паповавирусы (прогрессирующая многоочаговая лейкоэнцефалопатия), вирус краснухи (прогрессирующий панэнцефалит как последствие врожденной краснухи), цитомегаловирус (цитомегаловирусная инфекция мозга), вирус простого герпеса (подострый энцефалит), аденовирусы типов 7 и 32 (подострый энцефалит), вирус клещевого энцефалита и другие тогавирусы (эпилепсия Кожевникова и прогрессирующий бульбарный паралич). Продолжаются поиски этиологических агентов таких медленных заболеваний ЦНС, как рассеянный склероз, болезнь Паркинсона, шизофрения, болезнь Альцгеймера.
Просматривается и тенденция к расширению классической концепции о медленных инфекциях. По логике к ним могут быть отнесены не только дегенеративные изменения ЦНС, но и ряд других заболеваний, вирусная (или вирусоподобная) природа которых доказана или может быть установлена в будущем. Например, медленной инфекцией можно считать СПИД (это относится не только к ВИЧ-индуцированному поражению ЦНС), хронический гепатит В и С, ретровирусный Т-клеточный лейкоз. В опытах на животных обнаружено влияние вирусной персистенции на функции эндокринных желез (карликовость, диабет), подозревается участие вирусов в развитии атеросклероза и аутоиммунных заболеваний. Здесь кроме новых герпесвирусов большой интерес вызывают эндогенные ретровирусы, которые обнаружены в различных тканях, включая плаценту. Наиболее одержимые сторонники патогенетически значимой персистенции вирусов связывают с ней развитие опухолей и даже старение организма. Что ж, и наука нуждается в интригах.
Воспаление и инфекционный процесс
Существенный и первичный элемент воспаления состоит в реакции фагоцитов против вредных деятелей.
И.И. Мечников
- Воспалительная реакция: сано- и патогенез.
- Первичная и вторичная альтерация.
- Фагоциты в сетевых механизмах воспаления.
- Завершенное и незавершенное воспаление.
- Факторы транскрипции в регуляции воспалительных реакций.
- NF-kB и взаимоотношения с флогогенными стимулами.
- NF-kB в противовоспалительной терапии.
- Эффекторы воспаления и управление здоровьем.
Диалектика воспаления
Учение о воспалении возникло на основе клинических наблюдений. Утверждение общебиологического (иммунитетного) смысла воспалительных реакций потребовало долгих споров, хотя задним числом трудно понять упорство противников адаптивной сущности воспалительного процесса. Впрочем, и сегодня прагматизм, который был и остается базисом медицины, побуждает смотреть на воспаление с утилитарных позиций, трансформируя через них изучение фундаментальных проблем этого важнейшего аппарата гомеостаза.
Воспаление служит патогенетической основой инфекционных заболеваний. Оно возбуждается микробной инвазией и эволюционирует вместе с ним. Говоря о динамике воспалительного и одновременно инфекционного процесса (т.е. об их развитии во времени, от начала до завершения или перехода в осложненные формы), мы исходим из принципа, что нет неадекватных реакций, а есть неадекватные раздражители. При этом подразумевается, что неадекватность раздражения — понятие относительное, отражающее как степень самого раздражения (например, качественные и количественные параметры инфекции), так и способность организма нейтрализовать его. Слабое повреждение вызывает слабую реакцию без внешних, клинически значимых признаков. Такого рода латентные (субклинические) формы, возникающие при внедрении в организм малых дестабилизирующих агентов, можно рассматривать как физиологическое воспаление или микровоспаление. Чрезвычайное повреждение требует чрезвычайных мер — патологического воспаления. О его тенденциях судят по клиническим симптомам, т.е. по внешне заметной динамике. Быстрая ликвидация причины, побудившей реакцию, характерна для острого воспаления. Перманентное раздражение вызывает хронизацию (стабилизацию) процесса.
Чтобы понять диалектику воспаления и сопряженной с ним патологии, необходимо знать движущую силу воспалительного процесса. Речь идет не о частных детерминантах, а о наиболее общем, базисном механизме, инициирующем и поддерживающем воспалительные реакции. Возникая в ответ на повреждение, он нацелен на восстановление гомеостаза (т.е. динамического постоянства внутренней среды), развиваясь или подвергаясь обратному развитию вместе с решением этой задачи. Повреждение в той или иной форме присутствует на всех этапах воспаления, оставаясь главным мотивом, который организует и направляет развитие процесса. Отсюда символическая формула: эволюция воспаления есть эволюция повреждения.
Кроме повреждения, воспалением управляет стремление к удалению (элиминации) флогогенных начал и к репарации, т.е. восстановлению поврежденных тканей. Впрочем, все эти три события (повреждение, элиминация, репарация) разобщены лишь формально. В действительности они взаимосвязаны в каскадных (точнее, сетевых) реакциях, не только следуя одно за другим, но и взаимообусловливая друг друга. Можно сказать, что воспаление — это суперпроцесс. Его развитие определяется совокупностью (а главное — соизмеримостью) многих факторов и механизмов, хотя именно повреждение, инициируя реакцию, остается ее главной движущей силой. Элиминация и репарация работают на восстановление гомеостаза, но, решая эту задачу, сами включаются в повреждение.
Первичная и вторичная альтерация
Флогогенная (в том числе инфекционная) альтерация складывается из двух основных компонентов — первичного и вторичного повреждений. В первом случае речь идет о действии пускового (этиологического) фактора, вторичное — возникает как ответ на первичную альтерацию. Оно сопряжено с механизмами, которые нацелены на ликвидацию пускового агента, но, стремясь к этому, усиливают нарушения гомеостаза, повышая исходную флогогенность. В этой схеме заложен глубокий патогенетический смысл: расчленение действия первичных флогогенов и реактивной альтерации, опосредуемой эффекторами воспаления. Реактивный характер вторичной альтерации отличает ее от первичного поражения, которое всегда пассивно. Вторичное повреждение формируется в масштабе гуморально-клеточной и межклеточной кооперации крови и соединительной ткани, при дестабилизации которых вычленяются медиаторы воспалительного процесса, в том числе эффекторы повреждения — вторичные флогогены, или (применительно к инфекционной патологии) вторичные токсины.
При экссудативно-деструктивном воспалении в центре стоит нейтрофил с быстро реализуемым, высокоагрессивным потенциалом. Это определяет острое развитие событий с тенденцией к быстрому разрешению. При пролиферативно-деструктивном (гранулематозном) воспалении процесс с самого начала обретает хроническое течение. В первом случае главным механизмом является нейтрофилзависимое повреждение эндотелия, во втором — макрофагиндуцированный фиброгенез. Сетевой характер воспаления ведет к подключению новых факторов, представляющих разные уровни гомеостаза. Наращивание эффекторного каскада и одновременное расширение спектра мишеней продолжается до тех пор, пока не будут удалены первичные и вторичные флогогены либо во вновь возникшей системе не сложится состояние относительного равновесия. О нем можно говорить как о патологическом (или адаптивном) гомеостазе, который формируется при любых хронических процессах, а при инфекционных заболеваниях отражает коадаптацию хозяина и паразита.
Многофакторность не должна заслонять главных эффекторов воспалительной реакции — фагоцитов. Именно от них во многом зависит удаление флогогенных агентов и реактивное повреждение, связанное с гиперболизацией элиминационного процесса. Все остальное — своего рода ассистирующий персонал, который подготавливает либо завершает работу фагоцитов. Вместе с тем причины клинической неадекватности воспалительной реакции (в ее гипо- или гиперреактивных вариантах) могут быть расшифрованы только в масштабе всего гуморально-клеточного каскада. Если клинически значимое воспаление символизирует чрезвычайное раздражение, которое не может быть ликвидировано в рамках физиологических реакций, то осложненное течение говорит о еще большем несоответствии между повреждающими и санирующими механизмами, о преобладании первых над вторыми или, в лучшем случае, об их относительном равновесии.
В учении о реактивности есть правило исходного состояния. Его суть в том, что интенсивность реакции на один и тот же стимул зависит от функционального состояния (реактивности) отвечающей системы, или, говоря словами А.Д. Сперанского, сила раздражителя есть степень раздражения. Чувствительность фагоцитов к активирующим воздействиям тоже колеблется. Ее повышение носит название праймирования (от англ. prime — подготавливать) и возникает под влиянием факторов, привносимых извне либо генерируемых внутри организма, в системе гуморально-клеточной кооперации. Обладая повышенным эффекторным потенциалом, праймированные фагоциты лучше справляются со своими гомеостатическими функциями, но одновременно усиливают и патогенетические ресурсы, с которыми связана воспалительная альтерация тканей. В этом смысле праймирование не только инструмент, адаптированный к задачам саногенеза, но и потенциальный механизм гиперреактивного повреждения. Это означает, что в зависимости от ситуации следует стремиться либо к праймированию фагоцитов (например, для усиления противоинфекционной резистентности), либо, наоборот, к снижению их реактивности — для профилактики и терапии флогогенных осложнений. Более подробно эти вопросы прокомментированы ниже.
Воспаление и резервы иммунитета
Итак, компенсаторная сущность воспаления является следствием фагоцитарных реакций; неполноценность воспаления сопряжена с дефектами или нерациональным использованием фагоцитарных резервов. Клинически значимое воспаление — символ абсолютного или относительного неблагополучия эффекторов иммунитета, создающих несоответствие между гомеостатическими возможностями организма и повреждением. При абсолютном несоответствии пусковой фактор обладает силой, которая исключает вероятность его элиминации в рамках физиологических реакций. Такие ситуации возникают при заражении высоковирулентными штаммами бактерий или при внедрении массивной дозы инфекта. Относительное неблагополучие связано с ослаблением общей или местной резистентности, отражая неспособность к адекватным реакциям на обычное раздражение. Это характерно для вторичных (приобретенных) иммунодефицитов, требуя прогностической оценки и коррекции. Можно сказать, что если для физиологического (компенсаторного) воспаления имеется лимит раздражения (с ним связано понятие «норма реакции»), то для патологических реакций такого ограничения не существует. Они возникают при избытке повреждающих начал и определяются соотношением между механизмами, которые дестабилизируют и/или восстанавливают гомеостаз по ходу реакции.
Внутренняя среда служит местом постоянной инвазии условно-патогенных бактерий. В нормальном организме такая инфекция, сталкиваясь с факторами иммунитета, координируемыми воспалительной реакцией, обычно не получает развития. Нарушения реактивной защиты и ее резюмирующего звена, фагоцитоза, косвенно усиливают агрессивность инвазии, создавая предпосылки для реализации болезнетворности инфекционных агентов. Классические понятия «завершенный и незавершенный фагоцитоз» можно перенести на воспаление, отдача которого пропорциональна резервам защиты. При их снижении реакция буксует, персистируя вместе со стабилизацией инфекта (см. таблицу). Это пример, когда воспаление, будучи главным рычагом иммунитета, отражает не его силу, а слабость.
Факторы, способствующие хронизации (стабилизации) воспаления
|
Факторы |
Механизм действия |
|
Общие факторы |
|
| Прямые дефекты фагоцитов | Ослабление эффекторного звена иммунитета |
| Опосредованные дефекты: | |
| опсонопатии (дефекты комплемента, антителообразования и пр.) | То же |
| дефекты хемоаттрактантов (дефекты комплемента и других систем, генерирующих ранние хемоаттрактанты; избыток ингибиторов) | То же |
| дефекты эндотелиоцитов (слабая экспрессия адгезивных молекул) | То же |
| Прямые и опосредованные дефекты макрофагов и фибробластов | Ослабление репаративного звена воспаления |
|
Местные факторы |
|
| Анатомические дефекты | Нарушение дренажной функции воспаления |
| Ослабление мукоцилиарного транспорта | Усиление воспалительных эффектов |
| Биотоксические эффекты флогогенов | Ослабление элиминационных и репаративных механизмов воспаления |
| Склерозирование базальных мембран микрососудов | Подавление мобилизации эффекторов воспаления |
| Реинфицирование | Усиление воспалительных эффектов |
Клинический профиль подобных состояний определяется повышенной чувствительностью к возбудителям так называемых оппортунистических инфекций. При снижении ресурсов острого воспаления их причиной служат пиогенные бактерии, поражающие ослабленных (больных) людей. Иными словами, такие инфекции всегда вторичны, т.е. сопряжены с гипорезистентностью организма, осложняя течение базисного патологического процесса. При глубоких (как правило, врожденных) дефектах фагоцитоза воспаление трансформируется в затяжную, нередко полиорганную патологию, так как базисный механизм иммунитета ослаблен и не обеспечивает адекватного сопротивления возбудителям. В связи с тем, что бактерии внедряются через внешние покровы, дефекты фагоцитоза ведут к поражению слизистых оболочек и кожи; глубокие очаги наблюдаются редко.
Гипорезистентность может быть связана с местными факторами. В этом случае воспаление развивается как одиночный хронический очаг инфекции в естественных полостях или поврежденных тканях (например, раневая инфекция). В известном смысле такой процесс живет по законам собственного гомеостаза, который базируется на относительном равновесии механизмов повреждения, элиминации и репарации. Этому способствуют разные обстоятельства: анатомические пороки, нарушающие дренажную функцию (т.е. выведение дестабилизирующих агентов), вторичная слабость фагоцитов и фибробластов, испытывающих негативные воздействия бактериальных и других продуктов и т.д. Происходит то, о чем говорилось выше: не справляясь со своими задачами, реакция стабилизируется, обретая затяжной характер. Срабатывает принцип порочного круга, чему содействует и сокращение притока эффекторов (клеток, гуморальных факторов) на фоне склерозирования базальных мембран эндотелия и вторичного запустевания кровеносных сосудов.
Пиогенная инфекция может эволюционировать и как генерализованный (системный) процесс. Это обычно сопряжено с прорывом местного барьера, отражая его несостоятельность. В таком толковании есть очевидная, но не исчерпывающая логика. Даже при глубоких дефектах нейтрофильного фагоцитоза повышенная чувствительность к пиогенной инфекции редко сочетается с системными осложнениями. Это связано с сохранением бактерицидности плазмы и элиминационных ресурсов резидентных макрофагов ретикулоэндотелиальной системы, которые в кооперации с опсонинами очищают кровь от чужеродных агентов. Действительно, лишь при совместных дефектах поли- и мононуклеарных фагоцитов обнаруживается склонность к абсцессам внутренних органов, остеомиелиту и другим проявлениям вторичного заноса инфекции в глубокие ткани.
Усиление эффекторов воспаления
Свое первое сообщение о фагоцитарном иммунитете И.И. Мечников назвал «О целебных силах организма», обозначив идею управления здоровьем через фагоцитарные механизмы. Потребовались десятилетия, чтобы от деклараций перейти к реалиям, сменив эмпиризм на научные подходы к регуляции воспалительных реакций. О воспалении можно говорить как о защите, переходящей в повреждение. В этой формуле отражена диалектика воспалительного процесса, где всегда присутствуют элементы «пользы» и «вреда». Вред нацеливает на подавление реакции, польза требует ее усиления. И того и другого можно добиться, воздействуя на эффекторы воспаления, в том числе на фагоциты.
Идея об усилении фагоцитоза как способа управления здоровьем возникла тотчас после признания важнейшей роли фагоцитов. Поскольку иммунитет ассоциировался с устойчивостью к инфекциям, именно антимикробные свойства фагоцитов вызывали наибольший интерес. Развитие представлений о ретикулоэндотелиальной системе и ее ключевой позиции в очищении (клиренсе) внутренней среды от чужеродного материала вскрыли универсальность механизмов, связанных с фагоцитарными клетками. Утверждение за фагоцитами центральной позиции в реакциях иммунитета укрепило стремление к фармакотерапии через фагоцитарную стимуляцию. Полноценность фагоцитарных реакций зависит от комплекса гуморальных факторов — хемоаттрактантов, опсонинов, медиаторов, которые повышают реактивность фагоцитов, настраивают их на взаимодействие с эндотелиальными клетками и соединительнотканным матриксом. Это определяет новые пути усиления фагоцитарной защиты организма в целом. Все они стыкуются через воспаление — процесс, где проверяется надежность иммунитета, т.е. соответствие его эффекторов той агрессии, которую требуется погасить.
Фагоцитоз относится к неспецифическим механизмам иммунитета, так как фагоциты не дифференцируют объекты по антигенам. Но он имеет отношение и к реализации специфической защиты, используя для этого два канала. Один из них контролируется опсоническими антителами. Они обеспечивают избирательность фагоцитарных реакций, фокусируя их на антигеннесущих мишенях. Второй — связан с усилением цитотоксичности фагоцитов (прежде всего макрофагов) под влиянием цитокинов, секретируемых при антигенной стимуляции Т-лимфоцитами (Th1). Cопряженность различных механизмов иммунитета сглаживает остроту вопроса об избирательности иммунокорригирующих воздействий. Обычно это недостижимо и, по-видимому, не требуется. Вместе с тем, каким бы способом ни пытались поднять устойчивость организма, конечный итог зависит от эффекторного звена иммунитета, т.е. от тех факторов, которые непосредственно участвуют в элиминации патогенов. С этой точки зрения усиление фагоцитоза и готовности к воспалительной реакции следует признать одним из действенных критериев иммунокоррекции. Искусственная задержка (на 2—4 ч) эмиграции нейтрофилов в место введения пиогенных бактерий ведет к расширению зоны поражения, способствуя генерализации процесса. После проникновения менингококков в спинномозговой канал воспалительная реакция отсутствует в первые 12 ч. Это создает возможность опережающего размножения бактерий.
В 1940-х гг. В.И.Иоффе была предложена проба на «общую иммунологическую реактивность». Она учитывала местную реакцию при внутрикожном введении антител против тканей человека. Многочисленными исследованиями доказана эффективность метода в оценке клинических вариантов различных заболеваний, прогнозировании их исходов и осложнений. Конкретизация знаний привела к отрицанию универсализма в иммунодиагностике и породила скепсис к обобщающим понятиям.
Между тем при правильной трактовке проба В.И. Иоффе представляет несомненный интерес. Фактически она выявляет готовность к острой воспалительной реакции, которая развивается на инъекцию цитотоксической сыворотки и во многом зависит от реактивности нейтрофилов. Можно сожалеть, что этот простой прием, наделенный глубоким смыслом, не закрепился в практической медицине в своей первоначальной или модифицированной форме.
Впрочем, в более современных работах можно найти известные параллели. Так, интенсивность реакций на внутрикожное введение антигенов широко распространенных микробов (стрептококка, микобактерий туберкулеза, кандид и пр.), которые принято связывать с естественной сенсибилизацией Т-лимфоцитов, зависит и от мобилизационной активности нейтрофилов. Готовность к воспалительному ответу отражает и внутрикожная проба с фитогемагглютинином (ФГА). Считалось, что здесь также моделируется гиперчувствительность замедленного типа, так как ФГА принадлежит к мощным активаторам Т-лимфоцитов. Но он возбуждает и макрофаги. Поэтому ФГА-индуцированные реакции правильнее рассматривать как эквивалент мононуклеарного воспаления в целом, а не только как аналог антигензависимого процесса.
Стремление к точной локализации дефектов иммунитета не исключает, а, напротив, подчеркивает необходимость в обобщенных критериях, подтверждая правило: в биологии целое всегда больше суммы составляющих его частей. К показателям подобного типа относятся и воспалительные реакции. Развиваясь по каскадному принципу, воспаление может оборваться на любом этапе, если принятые меры способны ликвидировать повреждение. Но какие бы формы ни принимал воспалительный процесс, он остается очагом централизации и взаимодействия неспецифических и специфических эффекторов иммунитета. Именно здесь они проявляют свою силу или слабость, предупреждая повреждение либо включаясь в его развитие.
Комментируя приоритетность функциональных перестроек, инициируемых при воспалении в сети гуморально-клеточных и межклеточных взаимодействий, следует иметь в виду важность ответа на вопрос, где искать источник флогогенных цитокинов и других воспалительных медиаторов, и учитывать скорость мобилизации эффекторного потенциала, заложенного в отдельных клетках, включая фагоциты. Наряду с тучными клетками, ведущая роль здесь отводится макрофагам, которые в числе первых реагируют на нарушения структурной гармонии, за что даже получили название клеток тревоги. Патологам известен феномен тахифилаксии — кратковременного повышения резистентности к дестабилизирующим агентам после малых (компенсируемых) стресс-воздействий. Примечательно, что тахифилактогенной активностью обладают препараты, известные как стимуляторы ретикулоэндотелиальной системы — бактериальные липополисахариды, зимозан, дериваты пептидогликана и пр. Не исключено, что именно макрофаги, являясь мишенью для индукторов тахифилаксии, выступают и как ее пусковые эффекторы, усиливающие резервы адаптации. Достаточно вспомнить о цитокининдуцированном синтезе гепатоцитами острофазовых белков, играющих важную роль в адаптивных перестройках организма, сопряженных с воспалением (см. «Механизмы противоинфекционного иммунитета»). Три цитокина (интерлейкины-1, -6, туморонекротический фактор) в комплексе с глюкокортикоидами наиболее активны в возбуждении острофазового ответа. Их опережающим источником служат макрофаги.
Медицина пережила увлечение многими неспецифическими стимуляторами (пиротерапия, протеинотерапия, токсины Коля и пр.). Некоторые из них до сих пор фигурируют в фармакологических справочниках, хотя и потеряли былую популярность. На смену выходят новые препараты и процедуры, претендующие на оригинальность лечебного эффекта, но скорее всего повторяющие прошлое (озонотерапия, лазерная терапия, ультрафиолетовое облучение крови, новые варианты пиротерапии и пр.). Не исключено, что это та же стресс-терапия с элементами тахифилактогенности. Об этом следует помнить, учитывая универсальный эффект многих стимулов, нацеленных на индукцию клеточных транскрипционных факторов, поддерживающих воспаление (см. ниже).
Несколько замечаний по поводу опсонинзависимой регуляции фагоцитоза. Напомним, что к опсонинам относятся гуморальные факторы, которые, связываясь с объектами, усиливают их поглощение фагоцитами. Различают специфические (антитела) и неспецифические (комплемент, фибронектин и др.) опсонины, которые дополняют друг друга, вступая в функциональную кооперацию. Примером такой кооперации является взаимодействие антител (IgG и IgM) и комплемента (C3-фактор) с образованием мощного тандема, ассистирующего в уничтожении бактерий.
Опсонинам (особенно антителам) принадлежит ключевая роль в пластичности фагоцитарного иммунитета, что делает их реальным инструментом для управления воспалительными реакциями. Большинство опсонинов присутствует в нормальной плазме, которая в виде цельного препарата и отдельных фракций может быть использована для усиления фагоцитоза и коррекции опсонопатий. Коммерческие иммуноглобулины содержат богатый набор антител-опсонинов, что, безусловно, имеет отношение к их эффективности. Это особенно важно в лечении и профилактике пиогенных инфекций, устойчивость к которым почти целиком зависит от опсонинопосредованных реакций нейтрофилов. В связи с этим представляется целесообразным тестировать иммуноглобулины на опсоническую активность, учитывая этот признак в подборе дозировок терапевтических средств.
Между тем, в назначении иммуноглобулинов до сих пор доминирует эмпиризм, не подкрепленный объективными (иммунологическими) показаниями и критериями эффективности. Одним из них могло бы стать опсонинзависимое усиление фагоцитоза. На это можно рассчитывать при внутривенном введении достаточно больших объемов препарата, следуя не только классическим принципам заместительной терапии, но и учитывая взаимопотенцирующие эффекты в системе опсонической кооперации. Это же определяет целесообразность инстилляции иммуноглобулинов непосредственно в очаги воспаления в расчете на кооперацию с экссудативными фагоцитами и исправление дефектов местных опсонинов.
Для коррекции комплементзависимых опсонопатий специфические препараты отсутствуют, хотя позитивный опыт применения фракций плазмы, обогащенных факторами комплемента, имеется. Добиться клинической ремиссии при врожденных опсонопатиях удавалось путем введения больших количеств свежей плазмы. Сегодня это единственный способ воздействия на глубокие дефекты комплемента, которые, к счастью (как и другие врожденные пороки иммунитета), встречаются редко.
Открытие опсонических свойств фибронектина явилось основанием для испытания обогащенных им препаратов. Фибронектин плазмы взаимодействует главным образом с фиксированными макрофагами, способствуя очищению крови от продуктов деградации коллагена, фибриновых сгустков, тромбопластина, тромбоцитарных агрегатов, протеолитических ферментов, иммунных комплексов и других факторов, провоцирующих или поддерживающих внутрисосудистую активацию крови (своего рода «воспаление изнутри»). Многие патологические состояния (травмы, ожоги, интоксикация, ДВС-синдром и пр.) сопровождаются повышенным расходом фибронектина, определяя целесообразность заместительной терапии, в том числе в масштабе антистрессовых мероприятий. Для этого предложено использовать криопреципитат плазмы — субстрат, обогащенный многими факторами, в том числе фибронектином. Его введение оказалось полезным при тяжелых травмах, ожогах, септической интоксикации. Есть данные, что клиническое улучшение сочетается со стимуляцией клиринговой функции печени, повышением уровня плазменного фибронектина, усилением опсонической активности сыворотки. Наряду с этим ряд авторов с сомнением относится к применению криопреципитата и прежде всего к трактовке его действующего начала. Одно из возражений связано с низкой дозировкой вводимого фибронектина, которая не позволяет добиться полноценного заместительного эффекта, особенно у взрослых. Опыт клинического испытания очищенного фибронектина дал невыразительные результаты.
Своеобразной имитацией макрофагального фильтра крови являются экстракорпоральные гемосорбенты. Извлекая из циркуляции токсичные (дестабилизирующие) продукты, они усиливают естественную элиминацию агрессивных факторов, потенцируя опсонофагоцитарные механизмы. С этой точки зрения приемы эфферентной терапии следует отнести к разряду фагоцитоззамещающих методов лечения. Однако и здесь формальная логика заводит в тупик. Объем гемосорбентов, как правило, слишком мал, чтобы их эффект целиком проецировать на очищение крови от дестабилизирующих агентов. Определенную результативность дают эндогенные медиаторы (производные комплемента, цитокины и пр.), которые, образуясь при контакте крови с чужеродной поверхностью, запускают каскад стрессзависимых реакций. При использовании биологических сорбентов (например, гемоспленосорбция) стимулирующие агенты могут вымываться из самого сорбента, дополняя элиминирующий эффект операции. Это вносит в процедуру элементы тахифилаксии с ее не только позитивными, но и негативными последствиями. Во всяком случае, этого трудно избежать, если не прибегать к специальным мерам для снижения реактогенности гемосорбентов.
Как уже говорилось, возбуждение фагоцитов может происходить по типу субактивации или праймирования, когда о функциональной перестройке клетки удается судить не по внешней реакции, а по изменению чувствительности к повторным стимулам. Способность к праймированию определяет возможность усиления еще не начавшихся фагоцитарных реакций, т.е. повышения готовности клеток к предстоящему выполнению своих функций. Идеальный (а потому вряд ли достижимый) вариант пластичности фагоцитарных реакций — профилактическое праймирование в группах риска, например по оппортунистическим инфекциям. На практике чаще имеют дело с больными, усиливая защиту на фоне сформировавшихся патологических процессов. В таких случаях эффекторы гомеостаза уже испытывают напряжение, и вопрос о дополнительной стимуляции необходимо решать, учитывая сохранившиеся резервы защиты. Судя по состоянию нейтрофилов и моноцитов крови, даже при тяжелых инфекциях и воспалительно-деструктивных процессах фагоциты могут не только сохранять, но и усиливать свой реактивный потенциал. Безусловно, всему есть предел, и в наиболее тяжелых (прогностически неблагоприятных) случаях реактивность клеток падает.
Представления о резервах функциональной емкости фагоцитов связаны с вопросом о принципах их стимуляции. Можно ли, к примеру, усилить клетки непосредственно в очаге воспаления или это надо делать с опережением — в кровяном русле или даже на уровне костномозговых предшественников? Иными словами, можно ли повысить эффекторную отдачу очага воспаления за счет дополнительной стимуляции уже вступивших в реакцию фагоцитов или необходим приток свежих клеток? Безусловно, многое зависит от конкретных обстоятельств, прежде всего от степени раздражения фагоцитов. Но уместно говорить и об универсальных закономерностях, которые лишь недавно получили отражение в клинических работах. Наиболее общий вывод сводится к тому, что фагоциты сохраняют способность отвечать на рестимулирующие воздействия, даже отработав в том же функциональном режиме, например, в хемотаксических и бактерицидных реакциях. Это говорит о том, что, подобрав адекватные стимулы, можно усилить начавшийся фагоцитарный процесс, а, следовательно, и эффекторную отдачу воспалительной реакции. Необходимо лишь контролировать нагрузку, которую клетка несет от основного раздражителя.
Много неясного остается в вопросах заместительной терапии клеточных дефектов фагоцитоза. Это прежде всего касается практической стороны дела, так как логика самой процедуры очевидна.
Наиболее доступный субстрат для заместительной терапии — лейкоцитарная масса. При введении лейкоцитов в кровь рассчитывают на эффект от поступления дополнительной порции клеток (главным образом нейтрофилов) с антимикробным действием. Однако вливание лейкоцитов (кроме заместительного эффекта) неспецифически стимулирует резистентность, активируя механизмы тахифилаксии. Этим можно объяснить положительное действие системных лейкоцитарных инфузий даже в тех случаях, когда количество и функциональное состояние вводимых клеток не позволяют рассчитывать на заместительный эффект. Альтернативой являются местные аппликации лейкоцитарной массы с прицелом на ликвидацию изолированных очагов пиогенной инфекции. В целом трансфузия лейкоцитов — не рутинная процедура, а дорогостоящий метод с элементами риска для донора и реципиента. Наиболее общее показание к переливанию лейкоцитарной массы — нейтропения (менее 1000 нейтрофилов/мл) на фоне истощения костномозгового резерва (апластическая анемия, длительная антиметаболитная терапия).
Кроме лейкоцитов предложено использовать макрофаги, получаемые при культивировании аутологичных моноцитов. Их местные аппликации дали обнадеживающие результаты у больных с гнойно-деструктивными бронхитами, гайморитами, нагноившимися ранами, при туберкулезе легких, осложненном неспецифическим воспалением. Технические сложности метода очевидны, а потому его применение не вышло за рамки авторских испытаний.
Сегодня предпочитают введение гемопоэтических цитокинов (например, колониестимулирующего гранулоцитарно-макрофагального фактора), воздействующих на стволовые клетки костного мозга. Они обеспечивают нарастание гемопоэза без неясностей, связанных с инъекциями чужеродных лейкоцитов.
Проблемы противовоспалительной терапии
Ослабление фагоцитарных реакций сопряжено с противовоспалительной терапией, издавна практикуемой при хронических заболеваниях. Ее стратегию можно определить как снижение вероятности возбуждения фагоцитов, нацеленное на ослабление их патогенетического потенциала. Сюда относятся применение этиотропных средств (при инфекционном воспалении), подавление образования и инактивация флогогенных медиаторов, блокада стимулирующих сигналов на уровне рецепции. Важнейшей точкой приложения противовоспалительных средств служит система «эндотелий—лейкоциты», где решаются ключевые вопросы, связанные с мобилизацией клеток в очаги воспаления. Крупным событием явилось раскрытие молекулярных основ адгезивных контактов лейкоцитов с эндотелиальными клетками и матриксными белками соединительной ткани. Современные представления о молекулах контактного взаимодействия (интегринах, селектинах и пр.) и регуляции их экспрессии определяют новые подходы к управлению воспалительными реакциями, позволяя понять особенности формирования клеточных инфильтратов при различных вариантах воспаления и своеобразие функциональной переадаптации клеток в зонах действия флогогенных стимулов.
Противовоспалительная активность может быть суммой нескольких механизмов, отражая сходство эффекторов на разных этапах воспалительного процесса, а также поливалентное действие препаратов. К примеру, глюкокортикоиды подавляют макрофагальный синтез флогогенных цитокинов и одновременно блокируют экспрессию адгезивных молекул на эндотелиоцитах. Сложных эффектов можно ожидать от нейтрализации цитокинов и других флогогенных молекул. Они имеют несколько (иногда множество) мишеней, чувствительность которых колеблется в динамике воспалительной реакции.
Сегодня следует говорить еще об одном открытии, которое имеет отношение к клеткам воспалительных реакций. Речь идет о важном рычаге клеточной транскрипции — нуклеарном факторе NF-kB — и сложной системе его адаптивной регуляции.
NF-kВ и воспаление
Подобно всем реакциям воспаление является адаптивным процессом, который требует привлечения суммы клеточных и гуморальных факторов. Многие из них продуцируются заново в ответ на воспалительные стимулы. Их образование требует синтеза молекул, нацеленных на возбуждение, развитие и терминацию реакции. Как индуктивное событие воспаление зависит от транскрипционных факторов, которые участвуют в регуляции клеточных генов, необходимых для воспалительного процесса.
Около 20 лет назад группа D. Baltimore обнаружила в мышиных В-лимфоцитах транскрипционный регулятор гена легкой k-цепи иммуноглобулинов, получивший название нуклеарного фактора kВ (NF-kB). Вскоре оказалось, что NF-kB присутствует во всех клетках взрослого организма, занимая центральные позиции в регуляции более 100 (по другим данным, более 300) генов, ответственных за индуктивный гомеостаз. Огромное число современных публикаций посвящено молекулярным механизмам активации NF-kB, его участию в реакциях неспецифического и специфического иммунитета, регуляции клеточного апоптоза и онкогенной трансформации, изысканию препаратов, контролирующих NF-kB-зависимые реакции.
Биохимия активации NF-kB. NF-kB представляет семейство цитоплазматических белков, которые находятся под негативным контролем, но при стимуляции переходят в свободное состояние, перемещаясь в ядро, где проявляют активность после связывания с промоторными участками генов. Это один из главных транскрипционных факторов, отвечающих за адаптивные клеточные реакции. Активацию NF-kB вызывают физические и химические агенты (радиация, ультрафиолетовый свет, повышенное давление, окислительный стресс, присутствие форболовых эфиров и др.), инфекционные факторы (бактерии, вирусы, паразиты и их продукты), сигнальные молекулы (гормоны, цитокины, ростовые факторы, цАМФ и др.). NF-kB влияет на различные гены, задействованные в иммунном, острофазовом и воспалительном ответах (см. рисунок). Сюда относятся цитокины (ИЛ-1b, ТНФ-a, ИЛ-2, ИЛ-6 и др.), хемокины (ИЛ-8, МСР-1, эотаксин и др.), индуцибельные ферменты (iNOS, Cox-2), адгезивные молекулы (ICAM-1, VCAM-1, Е-селектин), главный комплекс гистосовместимости (MHC-I, MHC-II), белки комплемента (В, С3, С4), ингибиторы и активаторы апоптоза (с-IAP1, c-IAP2, FasL, Bcl-2, TRAF-1, TRAF-2 и др.), факторы, контролирующие клеточный цикл (р53, циклин D1 и др.), белки острой фазы (С-реактивный белок, амилоид А и др.).


Логичным выглядит открытие NF-kB в нейтрофилах. Лишенные способности к пролиферации и резко ограниченные в синтетических возможностях, они привлекают NF-kB к регуляции собственного апоптоза, реакциям на микробные модулины, хемоаттрактанты, цитокины и другие факторы. Становится понятным, каким образом клетки, подобные нейтрофилу, отвечают на стимуляцию, повышая или снижая свой мобилизационный потенциал.
NF-kB представлен пятью белками: р50/105, р52/100, р65 (RelA), c-Rel и RelB. Их NH2-конец имеет Rel-гомологичный домен (он состоит из 300 аминокислот), который обеспечивает внутриядерный транспорт NF-kB, соединение с ДНК, ингибиторами и образование димерных молекул. Несмотря на множество форм, обычным типом NF-kB является гетеродимер р50-р65. Он присутствует в большинстве клеток и имеет необходимые сайты для связывания с генными промоторами. Гомодимеры р50 не активны, так как не содержат трансактивирующих доменов. Они выступают в роли репрессоров.
Экспрессия NF-kB отличается своеобразием. Например, синовиальные фибробласты активируют р50 и р65 для секреции ИЛ-6, тогда как тромбин побуждает эндотелиоциты к синтезу ICAM-1 через индукцию р65. Такая специфика дополняется особенностями регуляторных молекул (см. ниже). Наряду с разнообразием NF-kB-структур и обилием связывающих участков ДНК они обеспечивают чувствительность к множеству стимулов. Картина напоминает генетические регулоны бактерий, которые контролируются общими медиаторами, принимающими сигналы из внешней среды (см. «Болезнетворность бактерий»). Сюда следует добавить химические реакции, влияющие на функциональные свойства NF-kB: ацетилирование, фосфорилирование и S-нитротиолирование. Все это создает картину поливалентной активации NF-kB, которая допускает множество регуляторных (возможно, избыточных) путей к реализации эффекторного потенциала клеток.
IkB- и IKK-зависимая регуляция NF-kB. NF-kB присутствует в цитоплазме в виде неактивного комплекса с ингибиторами, IkB (см. рисунок). Семейство IkB-белков включает IkBa, IkBb, IkBg, IkBd, IkВe и фактор-3 В-клеточной лимфомы (Bcl-3). Все они имеют несколько так называемых анкириновых повторов (состоят из 30—33 аминокислот), связывающихся с Rel-доменом NF-kB. Активация вызывает фосфорилирование и убиквитинизацию IkB. Это меняет конформационную структуру молекул, определяя их разрушение внутри протеасом.
Лучше всего изучен IkBa. Он прочно связан с р65-субъединицей NF-kB, блокируя его активность. При стимуляции клеток происходит фосфорилирование IkBa (по 32 и 36 сериновым остаткам) и связывание (по 21 и 22 лизиновым остаткам) с белком-убиквитином (эффект убиквитиновой лигазы). Это вызывает протеолиз в системе 26S-протеасомного комплекса, который ведет к высвобождению NF-kB, мигрирующего в ядро, к месту своего действия. Транскрипционная активность NF-kB проявляется через считанные минуты после клеточной стимуляции.
Разные типы IkB оказывают неодинаковое или перекрывающее действие; их тканевое распределение тоже неравнозначно. Можно сказать, что клетки располагают набором IkB-молекул с разной чувствительностью к стимулам, неодинаковой кинетикой деградации, ресинтеза и аффинности. Это способствует дифференцированному поведению IkB в стимулированных клетках. К примеру, IkBa поддается быстрой инактивации, но столь же быстро восстанавливается. Это происходит из-за того, что свободный NF-kB связывается с промоторным участком IkBa-гена, вызывая синтез новых порций IkBa. Напротив, IkBb-ген не регулируется NF-kB. Поэтому вслед за деградацией IkBb наступает длительная NF-kB-активация.
Протеолиз IkB происходит в цитоплазме, но возможна и их ядерная деградация, например в нейтрофилах. Это должно способствовать оперативным событиям при остром воспалении. В таком случае следует допустить нуклеарную локализацию аппаратов убиквитинизации и протеасомного расщепления белков.
К деградации IkB ведет обилие сигнальных путей, но все они сходятся на активации IkB-киназного комплекса (IKK) (см. рисунок). Это гетеродимер, состоящий из двух белков IKK-a и IKK-b, скрепленных молекулой IKK-g, или NEMO (от англ. NF-kB essential modulator). IKK-комплекс активируется многими киназами, такими как изоформы протеинкиназы С (РКС); NIK, MEKK-1, MEKK-2, MEKK-3 и другие члены семейства МАРККК; NAK; фосфатидилинозитол-3-киназа и др. Их действие ведет к изменению эффекторных свойств IKK, нацеливая на фосфорилирование, взаимодействие с убиквитином и протеасомную деградацию IkB. Каналы, активирующие IKK, дополняются белками, передающими сигналы из рецепторного звена в систему IKK-киназ. К ним принадлежат TRAF-2 (от англ. TNF-receptor associated factor), TRAF-5, TRAF-6, MyD88 и др.
Главную роль в воспалении играет IKK-b; IKK-a больше занят в реакциях лимфоидных органов.
NF-kB и реализация воспаления. NF-kB принадлежит центральная позиция в управлении воспалительным процессом как мононуклеарного (например, замедленной гиперчувствительности), так и нейтрофильного (пиогенного) типов. Активация NF-kB повышает экспрессию адгезивных молекул, стимулирует трансэндотелиальную миграцию лейкоцитов, потенцирует образование воспалительных цитокинов, хемокинов и индуцибельных ферментов. В воспалительных клетках усиливается продукция IKK (прежде всего IKK-b), повышается ядерная локализация NF-kB.
Воспалительные гены могут регулироваться и другими транскрипционными факторами, например из семейства активационного белка, АР-1 и трех каскадов стимулирующей его киназы МАРК (митогенактивированная протеинкиназа): ERK (экстрацеллюлярно-регулируемая киназа), JNK (c-Jun-N-терминальная киназа) и р38 МАРК. Но важнее то, что МАРК-киназы способны сами фосфорилировать IKK с выходом на IkB-зависимую активацию NF-kB.
Признаки повышенной активности NF-kB обнаруживаются в очагах воспаления при многих заболеваниях человека: ревматоидный артрит, бронхиальная астма, хронические заболевания нервной системы, кишечника, атеросклероз, септический шок и др. Если учесть, что воспаление связано с апоптозом, следует напомнить о ключевых функциях NF-kB в запрограммированной смерти клеток. Активация NF-kB обычно задерживает апоптоз, продлевая жизнь клеток-эффекторов в зоне воспаления.
NF-kB и инфекционные агенты. Поскольку любая инфекция сопровождается воспалением, связанным с усилением транскрипции воспалительных генов, логичны поиски активированного NF-kB при инфекционных процессах. Действительно, бактериальные и другие инфекции вызывают активацию NF-kB, которая коррелирует со степенью воспаления. Первое впечатление, что для этого требуется инвазивная инфекция, оказалось неточным: активация NF-kB возможна с неинвазивными бактериями и их компонентами. Важным является функционально значимое взаимодействие с клетками, которое может ограничиться прикреплением к клеточным рецепторам или инъекцией микробных факторов внутрь клетки (это можно назвать молекулярной инвазией). Так ведут себя не только патогенные микроорганизмы, но и бактерии-пробиотики, т.е. «бактерии для жизни».
Некоторые возбудители используют активацию NF-kB для реализации своей патогенности. Полагают, что борьба с апоптозом, в которой участвует NF-kB, является для них полезным фактором. К подобным инфекциям относятся листериоз, хеликобактериоз, гонорея, брюшной тиф, шигеллезная дизентерия и др. Их возбудители способны развиваться внутри клеток, т.е. зависят от жизненного потенциала своих мишеней.
Но есть микробы, которые ведут себя иначе, блокируя NF-kB. Примером являются бактерии рода Yersinia. Они инъецируют в клетки факторы, подавляющие протеинкиназы (для этого они используют свой аппарат секреции III типа). Белки наружной мембраны (Yop) Y.рseudotuberculosis, Y.pestis и Y.enterocolitica атакуют ферменты митогенактивированной киназы (МАРК), которые, как сказано выше, участвуют в активации NF-kB. YopJ (Y. pseudotuberculosis) действует и как сериновая протеаза, инактивирующая убиквитин. Белок YopP (Y. enterocolitica) связывается с IKK-b, препятствуя активации NF-kB; это усиливает апоптоз макрофагов, ослабляя сопротивление инфекции. Другая стратегия направлена на подавление деградации IkB. Это происходит благодаря торможению фосфорилирования IkB, нарушению его убиквитинизации и блокировке протеасом. Так ведут себя уропатогенные штаммы E. coli и кишечные комменсалы, продуцирующие факторы, препятствующие высвобождению IkB из комплекса с NF-kB.
Некоторые бактерии действуют на более поздних сроках, инактивируя NF-kB после снятия с него IkB-блокады. Это наблюдается при связывании свободного NF-kB с микробными белками, которые имитируют анкириновые повторы IkB-ингибиторов. Нарушается фосфорилирование NF-kB — необходимый этап ядерной транслокации NF-kB. Так обстоит дело с Mycobacterium ulcerans, который подавляет активность NF-kB в Т-клетках и моноцитах, снижая напряженность иммунитета. Нечто подобное происходит при инвазии макрофагов простейшим паразитом Toxoplasma gondii. Он сдерживает ядерную транслокацию NF-kB, подавляя синтез провоспалительных цитокинов и развитие антитоксоплазменного иммунитета. Иначе обстоит дело с иерсиниями. Например, YopH (Y. рseudotuberculosis) проявляет свойства тирозинфосфатазы. Блокируя фосфорилирование свободного NF-kB, она снижает генетическую транскрипцию, ослабляя синтез воспалительных цитокинов и провоцируя гибель клеток.
Многие (если не все) вирусы активируют систему NF-kB. Это наблюдается у вирусов иммунодефицита человека, вирусов гепатита В и С, герпесвирусов, аденовирусов, вирусов гриппа, парамиксовирусов, риновирусов, ротавирусов и т.д. Они действуют через клеточные рецепторы (например, CD4 для вирусов иммунодефицита) или собственнные продукты (например, HBx вируса гепатита В, гемагглютинин ортомиксовирусов, E3-белок аденовирусов). Это важно для вирусов: затормозив апоптоз (за счет cтимуляции NF-kB), они продлевают свою репликацию. Но есть вирусы (например, вирус осповакцины), которые выступают в роли антагонистов NF-kB. Это усиливает апоптоз инфицированных клеток, обрывая вирусную инфекцию, т.е. скорее выгодно хозяину, чем паразиту.
Приведенные примеры демонстрируют взаимодействие едва ли не самого мощного экологического фактора (микробных агентов) с аппаратом воспаления. Это лишь первые исследования, но они открывают перспективу для изучения механизмов вирулентности и оказывающих им сопротивление эффекторов иммунитета.
Блокада NF-kB и противовоспалительная терапия. Многообразие медиаторных путей, возбуждающих NF-kB, показывает новые направления регуляции иммунных и воспалительных реакций. Главным является сам фактор NF-kB и его ингибиторы, IkB и IKK.
Перемещение NF-kB в ядро блокируется дитиокарбаматом пирролидина (ингибитор окислительного стресса) и глиотоксином (препарат из аспергилл). Блокаду активации NF-kB вызывают факторы, подавляющие серин-фосфорилирование, убиквитинизацию и протеасомный протеолиз IkB. Важно, что все они не влияют на способность белков IkB к комплексообразованию с NF-kB. Каскад реакций можно прервать и при помощи торможения IKK. Это задерживает активирующее разобщение комплекса NF-kB—IkB.
Для ингибиции NF-kB могут быть использованы генетические подходы — применение антисмысловых олигонуклеотидов; фрагментов ДНК, гомологичных NF-kB (decoy-факторов); IkB-cуперрепрессора, не поддающегося негативному фосфорилированию (IkB с двойной мутацией по серину 32 и 36). Следует помнить и о блокаде протеинкиназ, занимающих положение выше IKK (MAKP-киназы, PKC и др.). Это подтверждает их участие в активации IKK, а также в индукции других (дополняющих эффекты NF-kB) транскрипторов.
Большинство опытов проведено в клеточных культурах, но ряд наблюдений выполнен на животных. Примером служит подавление неспецифического и аутоиммунного воспаления при введении дитиокарбамата пиролидина, глиотоксина, NF-kB-decoy-ДНК-фрагментов, IkB-суперрепрессора или протеасомных ингибиторов. Все это пока не нашло практического применения или внедряется с осторожностью. Заметно опасение перед условной специфичностью большинства ингибиторов. Не ясно и то, что может случиться при длительном выключении NF-kB, если учесть его значимость для таких важных процессов, как пролиферация и апоптоз.
Тем не менее уже сегодня медицина использует ряд средств, действующих (по крайней мере частично) через торможение NF-kB. Так ведут себя глюкокортикоиды, которые прямо или косвенно блокируют активацию NF-kB. Из нестероидных препаратов следует отметить противовоспалительный эффект «малых» молекул, таких как аспирин, сульфасалазин, лефланомид и др. Их эффект ранее связывали исключительно с подавлением индуктивной циклооксигеназы-2 и синтеза противовоспалительных простагландинов. Но оказалось, что они активны и за счет блокирующего действия в системе NF-kB, в частности из-за подавления IkB-фосфорилирования.
Большой интерес вызывают ингибиторы деструктивной функции протеасом. Они обладают антионкогенным и противовоспалительным эффектами и направлены против активации NF-kB. С этой целью используют пептиды из группы борной кислоты типа PS-341 и PS-519. Такое же действие оказывают блокаторы убиквитиновой лигазы, переводящей IkB в объект для протеасомного протеолиза.
Ингибицию NF-kB могут вызывать и естественные продукты. Примером является куркумин — препарат из ризом Curcuma longa, растения южной и юго-восточной Азии. Куркумин (диферулоилметан) подавляет разрушение IkB, блокируя активацию IKK. Не случайно им давно пользуются как антивоспалительным средством в странах, где он производится в качестве вкусовой добавки. Интересный феномен обнаружен при изучении непатогенных кишечных бактерий. Авирулентные сальмонеллы и бактероиды при колонизации интестинальных эпителиоцитов подавляют убиквитинизацию IkB, снижая NF-kB-зависимое выделение цитокинов (похожее действие оказывает Saccharomyces boulardii, блокирующий сигнальные каналы МАРК). Лактобациллы, рекомендованные как пробиотик, выделяют факторы, вызывающие быстрый антипротеасомный эффект интестинальных эпителиоцитов, блокируя деструкцию ингибиторных факторов IkB. Все это позволяет говорить об участии NF-kB во взаимоотношениях эпителиальных клеток с нормальной микрофлорой и о возможности использования данного феномена в лечебных и профилактических целях.
* * *
Перечитывая И.И. Мечникова, удивляешься, насколько ясно ему представлялось будущее своей теории, в том числе ее прикладные аспекты. Многие из современных идей, связанных с регуляцией воспалительных реакций, были заложены И.И. Мечниковым и его ближайшими последователями. Эту преемственность не всегда просто разглядеть за изобилием фактов, новых терминов и изощренных методов. Но лозунг И.И. Мечникова «нет воспаления без фагоцитов», который (помня о диалектике воспалительных реакций) можно видоизменить: «Нет здоровья и болезней без воспаления и фагоцитов» — остается одним из главных направлений в борьбе за гомеостаз.
Апоптоз
Жить — значит умирать. Ф. Энгельс Ад и рай — две половины души.
О. Хайям
- Генетически запрограммированная смерть клеток.
- Альтернатива некротическому цитолизу.
- Индукция, регуляция и эффекторы апоптоза.
- Гомеостаз и патология.
- Взаимоотношения с инфекционными агентами.
Несмотря на непрерывное обновление, количество клеток в тканях взрослого организма остается постоянным. Это означает, что число вновь образующихся клеток соответствует числу отмирающих. Процесс физиологического отмирания клеток совершается незаметно, не сопровождается воспалительными реакциями, которые расширяют масштаб повреждения при некрозе.
Естественная смерть является вполне логичным событием, как любое замещение старого новым. Возможно, по этой причине интерес к нему просыпался медленно и лишь недавно достиг уровня концептуальных обобщений. Физиологическую смерть клеток стали называть апоптозом — термином, которым древние греки обозначали опадание лепестков с цветка или листьев с деревьев (греч. apoptosis — отделение, удаление). Классический апоптоз — не единственный механизм нормальной гибели клеток, но, безусловно, один из ее главных рычагов.
Понятие «апоптоз» введено в биологию в 1972 г., когда J. Kerr, A. Wyllie и A. Currie предложили его для характеристики комплекса однотипных морфологических изменений, которые наблюдаются при отмирании клеток, не связанном с некрозом. В их исследованиях признаки апоптоза были обнаружены в краевых зонах ишемических поражений; центральная область подвергалась некрозу. Современный интерес к этому феномену возник позже, в конце 1980-х гг., после того как понятие апоптоза слилось с представлениями о запрограммированной смерти клеток, информация о которой закодирована в клеточном геноме и поддается фенотипической регуляции. Было осознано, что апоптоз — такая же фундаментальная генетическая программа, как пролиферация, дифференцировка или пребывание клеток в покоящемся состоянии. Об апоптозе говорят как об активной смерти (противопоставляя его некрозу, т.е. пассивной гибели от «несчастного случая»), как о самоубийстве клеток, сознательно выбирающих дорогу смерти, если они чувствуют собственную бесполезность, а тем более опасность для клеточного сообщества. Общепризнано, что апоптоз занимает ключевые позиции в физиологии эмбрионального морфогенеза, метаморфозе, элиминации (негативной селекции) аутореактивных Т-лимфоцитов, уничтожении опухолевых и вирусинфицированных клеток. Широко обсуждается его роль в патологии — при аутоиммунных заболеваниях, злокачественных опухолях, дегенеративных и атрофических процессах, инфекционной патологии.
Феноменология апоптоза
Клетки, вступившие на путь апоптоза, претерпевают стереотипные изменения, ведущие к формированию характерного фенотипа (рис. 1, 2). Принципиально, что гибель наступает при сохранении целостности плазматической мембраны и внутриклеточных органелл. Поэтому клетка не разбухает (результат повреждения мембраны и переполнения клетки водой из окружающей среды), как в случае некроза, а сжимается, конденсируя цитоплазму и оставаясь непроницаемой для красителей типа трипанового синего или пропидий йодида. Клетки округляются, теряя микроворсинки, рецепторы и структуры, обеспечивающие клеточные функции и межклеточные контакты. Их контур становится гладким, и они отделяются от соседних клеток.

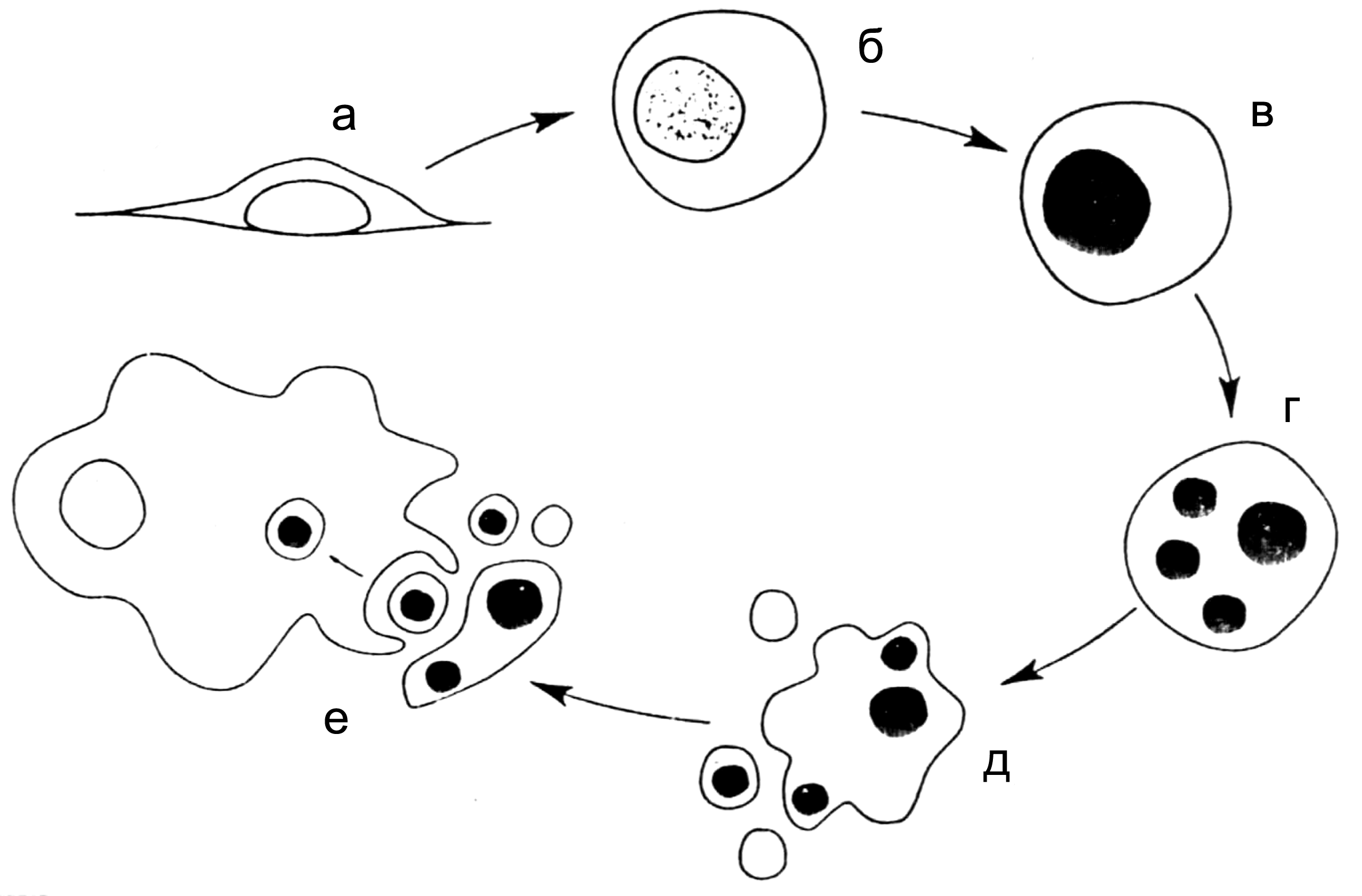

(препарат Н.А. Маянского)
Одновременно или раньше ядерный хроматин подвергается агрегации и расщепляется активированными эндонуклеазами на нуклеосомальные фрагменты, которые при электрофорезе разделяются на дискретные фракции (ДНК-лестница; рис. 3) в отличие от сплошного пятна деградированной ДНК некротизированных клеток (результат беспорядочного разрушения хроматина экзонуклеазами).
Вслед за этими изменениями (они совершаются в считанные минуты) в мембране образуются выпячивания, которые отпочковываются от клетки в виде апоптозных тел. Последние, как и сама клетка, окружены уплотненной (ригидной) мембраной и могут содержать органеллы и фрагменты ядра. Апоптозные тела поглощаются мононуклеарными фагоцитами, а также сбрасываются в окружающую среду. Поглощенный материал переваривается лизосомальными ферментами, причем макрофаги уничтожают его, не подвергаясь активации. Весь процесс, от агрегации хроматина до полного переваривания апоптозных тел, занимает 1—3 ч. Этим скоротечным событиям предшествуют латентные сдвиги, которые развиваются на протяжении 10—12 ч. На этом этапе клетки могут быть спасены от апоптоза продуктами протективных (антиапоптозных) генов; финальные события не обратимы.

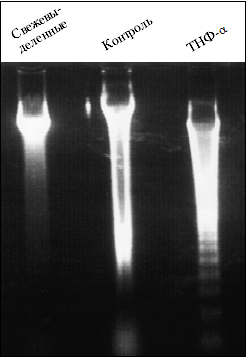
В целом морфологическая картина апоптоза отражает упорядоченность подключения механизмов, направленных на то, чтобы избежать взрывоопасных последствий цитолиза, когда из клетки высвобождается комплекс флогогенных начал, инициирующих воспалительную реакцию и сопряженные с ней вторичные повреждения. Однотипность цитопатических изменений позволяет думать об общности эффекторных механизмов апоптоза для любых клеток, по крайней мере на его заключительных этапах.
Методы регистрации апоптоза
Классический способ обнаружения апоптозных клеток основан на морфологических признаках, которые выявляются с помощью обычного светового микроскопа. Именно этот метод использовался для первого описания апоптозной гибели клеток и ее отделения от некроза. Клетки, вступившие на путь апоптоза, подвергаются характерным внешним изменениям (см. рис. 2): они уменьшаются в размерах (сморщиваются), ядерный хроматин уплотняется, ядро теряет свою структуру и распадается на несколько мелких образований (апоптозных тел). Подсчитав долю таких клеток в клеточной популяции, можно определить апоптоз количественно. Несмотря на свою простоту, морфологический метод достаточно трудоемок и субъективен. В настоящее время для исследования апоптоза имеется широкий набор других методик, хотя морфологический подход по-прежнему не утратил своего значения.
Для качественного определения апоптоза применяется электрофорез ДНК в агарозном геле. При анализе ДНК апоптозных клеток в геле регистрируется «апоптозная лестница» (см. рис. 3). Возникающая картина связана с тем, что при апоптозе ДНК расщепляется упорядоченно, т.е. между нуклеосомами (интернуклеосомное расщепление). Это ведет к накоплению наборов фрагментов ДНК, которые имеют разную электрофоретическую подвижность и формируют «лестницу». ДНК нормальных клеток в связи со своей большой массой остается в исходной точке, а при электрофорезе некротических клеток ДНК образует непрерывную полосу, отражающую беспорядочное расщепление и отсутствие наборов одинаковых фрагментов. Данный метод имеет сравнительно низкую чувствительность, но высокую специфичность.
При электрофорезе анализируется вся ДНК, экстрагированная из популяции клеток, что не позволяет проводить количественную оценку апоптоза. С этой целью используется другой метод, основанный на окрашивании ДНК с помощью пропидий йодида и последующем анализе на проточном цитофлюориметре. Пропидий йодид является специфическим красителем ДНК, так как при встраивании в молекулу двухцепочечной ДНК интенсивность его флюоресценции возрастает в 20 раз. Следовательно, свечение будет прямо коррелировать с содержанием ДНК в клетке. После обработки клеток гипотоническим раствором с добавлением детергента они набухают, и через дефекты в цитоплазматических мембранах могут выходить небольшие фрагменты ДНК, образовавшиеся при апоптозном расщеплении. При окрашивании пропидий йодидом такие клетки светятся менее ярко, так как имеют меньшее (гиподиплоидное) содержание ДНК по сравнению с нормальными клетками, в которых расщепление ДНК отсутствует (т.е. содержание ДНК — диплоидное). Этот метод прост в исполнении, достаточно чувствителен и специфичен.
При необходимости исследовать апоптоз в гистологических препаратах (in situ) применяется метод, также основанный на апоптозном расщеплении ДНК. Здесь используется то обстоятельство, что при апоптозном расщеплении ДНК образуется множество новых 3’-окончаний, к которым с помощью терминальной деоксинуклеотидилтрансферазы присоединяют меченые нуклеотиды (метод называется TUNEL от англ. TdT-mediated dUTP-nick end labelling). Несмотря на широкое применение ТUNEL-метода, его специфичность оспаривается. Имеются данные о том, что некротические и живые клетки во время репарации ДНК также окрашиваются положительно. В связи с этим были предложены дополнительные маркеры для исследования апоптоза in situ, например иммуногистохимическое окрашивание с помощью специфичных антител для выявления активных каспаз, в частности каспазы-3, которая активна только в апоптозных клетках. Вообще, активация каспаз служит важным признаком апоптоза и используется для его регистрации. Каспазная активность может быть определена количественно при расщеплении специфического субстрата каспаз. Эквивалентом активации каспаз служит появление в клетках их активных фрагментов, выявляемых путем иммуноблоттинга (англ. Western blotting) с антителами, специфичными для таких фрагментов.
При развитии апоптоза клетки изменяют свои поверхностные характеристики, экспрессируя мембранные структуры, которые отсутствуют у нормальных клеток. Наиболее популярным маркером служит фосфотидилсерин. Этот фосфолипид входит в состав внутреннего слоя клеточной мембраны и при апоптозе выныривает наружу, т.е. появляется во внешнем слое плазматической мембраны. Здесь он может быть выявлен с помощью белка аннексина-V, который в присутствии ионов Са2+ обладает высоким сродством к фосфотидилсерину. Клетки, меченые аннексином-V, конъюгированным с флюоресцентной меткой, анализируются с помощью проточного цитофлюориметра. Живые клетки не окрашиваются аннексином-V, тогда как взвесь апоптозных клеток будет аннексин-V-позитивной. Изменения в клеточной мембране относятся к ранним апоптозным событиям, поэтому этот метод может использоваться для изучения начальных проявлений апоптоза. К его преимуществам следует отнести возможность количественного определения уровня апоптоза, высокую чувствительность и специфичность.
Наиболее популярным методом оценки изменений биохимического профиля апоптозных клеток служит МТТ-метод, широко используемый для работы с опухолевыми клетками и оценки их выживаемости in vitro. Он основан на восстановлении красителя МТТ (3-[4,5-диметилтиазол-2-ил]-2,5-дифенилтетразолиумбромид) в лиловый продукт формазана под воздействием дегидрогеназ, активность которых в апоптозных клетках снижается. Реакция регистрируется колориметрически: чем больше живых клеток в культуре, тем больше будет образовываться формазана и больше меняться интенсивность окрашивания. Метод удобен для скрининга выживаемости клеток, поскольку может проводиться в микропланшетах с небольшим числом клеток. Однако следует подчеркнуть, что здесь определяется выживаемость клеток, и активность дегидрогеназ уменьшается не только в апоптозных, но и в некрозных клетках. Иными словами, не обязательно, чтобы «невыжившая» клетка погибла от апоптоза.
Таким образом, любой подход к регистрации апоптоза имеет свои преимущества и недостатки, поэтому всегда целесообразно начинать исследование с использования нескольких методов, определив корреляцию между ними, и впоследствии остановиться на наиболее удобном способе для решения конкретных задач.
Индукция апоптоза
По современным представлениям в каждой клетке имеются два основных механизма апоптоза: внешний (опосредованный через рецепторы смерти) и внутренний (митохондриальный, стресс-индуцированный). Большинство стрессирующих факторов (если они действуют в малых дозировках, не вызывая клеточного некроза) способны инициировать внутренний апоптоз. К ним относятся физические (радиация, ультрафиолетовое облучение, гипертермия) и химические (оксиданты, противоопухолевые препараты, токсины) стимулы, давно известные как цитотоксические агенты. Одним из универсальных индукторов апоптоза является окислительный стресс, который связан с реактогенными метаболитами кислорода, накапливающимися в клетках при различных воздействиях, особенно на фоне подавления антиоксидантных систем. Апоптоз служит стандартной реакцией на повреждение ДНК, и его индукция некоторыми агентами опосредована через этот канал.
В отличие от некроза апоптозный процесс развивается не только под влиянием формальных повреждений. Он возникает и как реакция на эндогенные факторы — гормоны, цитокины, дериваты арахидоновой кислоты, физические (прямые) межклеточные контакты. Общая формула такого «внешнего» апоптоза сводится к воздействию физиологических стимулов в нефизиологических концентрациях и сочетаниях. В подобных случаях апоптоз основан на рецепторном взаимодействии с индуцирующими агентами и зависит от типа клеток, их зрелости, физиологического состояния и природы стимулов. Удобной моделью для его изучения служат лимфоциты, которые подвергаются негативной селекции путем апоптоза, способны избегать апоптозной гибели, превращаясь в клетки памяти, а также индуцируют апоптоз в клетках-мишенях, добиваясь цитотоксического эффекта.
Имеются специальные рецепторы, сопряженные с активной смертью клеток. Важной является система Fas—FasL, где FasL cлужит лигандом, стимулирующим апоптоз после связывания с Fas (CD95), выступающим в роли рецептора. Fas имеется на многих клетках, тогда как FasL экспрессируется в основном активированными Т-лимфоцитами, являясь гомологом одного из важнейших цитокинов — туморонекротического фактора (ТНФ-a). Последний тоже относится к индукторам апоптоза и воспринимается рецепторами, по структуре напоминающими Fas. Цитоплазматический фрагмент Fas и ТНФ-рецепторов содержит домен смерти, транслирующий апоптозный сигнал (FasL, ТНФ-a) на внутриклеточный аппарат апоптоза. Этому механизму отводится важная роль в реализации гомеостатического и патогенетического потенциала цитотоксических CD8+ Т-лимфоцитов, или Т-киллеров.
Экстраполируя наблюдения на мутантных мышах, допускают, что дефекты в системе Fas—FasL способствуют развитию лимфопролиферативных и аутоиммунных заболеваний, а гипертрофия Fas-зависимого апоптоза чревата деструктивными осложнениями, в частности избыточным уничтожением вирусинфицированных клеток. Кстати, клетки иммунологической памяти, видимо, лишены Fas и, возможно, поэтому избегают уничтожения.
Альтернативой можно считать пассивный (спонтанный) апоптоз, или апоптоз «по умолчанию» (англ. default). Он не является результатом стимуляции клеток, а возникает при недостатке тонизирующих сигналов — ростовых факторов и других активаторов, препятствующих реализации программы клеточного самоубийства. Это могут быть классические гормоны или факторы, действующие по паракринному или аутокринному типу.
Пассивный апоптоз характерен для клеток с высоким дифференцировочным и пролиферативным потенциалом, которые нуждаются в постоянной стимуляции, поддерживающей их жизнеспособность и самообновление. Подобные ситуации типичны для эмбриогенеза, но возникают и во взрослом организме. Так, недостаток тестостерона и адренокортикотропного гормона ведет к апоптозной инволюции простаты и коры надпочечников. В инвитровых культурах элиминация ростовых цитокинов (таких как интерлейкин-3, гранулоцитарно-макрофагальный колониестимулирующий фактор, эритропоэтин) вызывает апоптоз стволовых клеток гемопоэза. Отсутствие интерлейкина-2 индуцирует апоптоз Т-лимфоцитов. Эндотелиальные клетки, моноциты и эозинофилы погибают соответственно без фибробластного ростового фактора, ТНФ-a, интерлейкина-5. Из наблюдений за клеточными культурами известно, что выживание отдельных клеток во многом зависит от функциональных и физических контактов с соседними клетками. Каждая клетка в отдельности настроена на апоптоз, но реализация или подавление программы смерти решаются клеточной популяцией в целом, подобно тому как регулируется и поддерживается баланс в масштабе целого организма.
Регуляция апоптоза
Если исходить из концепции об апоптозе как о конкретной генетической программе, логичным выглядит представление о специализированных генах, продукты которых усиливают или подавляют апоптозный процесс. В апоптозе задействовано около сотни молекул. Они связаны между собой в каскадных и сетевых взаимодействиях, дублируют друг друга либо демонстрируют уникальность позитивных и негативных эффектов, участвуют в физиологических актах или работают преимущественно на апоптоз.
Ростстимулирующие факторы нацелены на пролиферацию, апоптозные сигналы ведут к отмиранию клеток. Равновесие между обновлением и апоптозом обеспечивает баланс в клеточной популяции, перекосы создают предпосылки для гиперплазии или атрофии тканей. Большинство фактов, подтверждающих правильность данного рассуждения, получено в изящных экспериментах на нематоде Caenorhabditis elegans. Этот червячок состоит из 1090 соматических клеток; 131 из них отмирает по ходу его умирания. Установлена группа ced-мутаций (от англ. cell death), которые блокируют гибель клеток, приговоренных к смерти. У мутантных форм C. elegans такие клетки не только сохраняются, но даже участвуют в жизнедеятельности организма. Идентифицировано более десятка генов, регулирующих отдельные этапы апоптоза — начиная с принятия решения о самоубийстве до фагоцитоза и разрушения апоптозных клеток. Два гена, ced-3 и ced-4, необходимы для индукции клеточной смерти и, по-видимому, экспрессируются в апоптозных клетках. Ген ced-9 действует в противоположном направлении, подавляя апоптоз. Его мутация ведет к общей гибели организма, доказывая, что все клетки способны к апоптозу, но в норме он генетически заблокирован. Ряд генов определяют распознавание и поглощение апоптозных клеток соседними клетками, хотя непосредственно не вовлечены в процесс отмирания. Наконец, ген нуклеазы (nuc-1) контролирует ДНК-азную активность и необходим для разрушения фагоцитированного материала.
Изучение апоптоза у высших животных в принципе подтвердило концепцию о его дуалистической (позитивной и негативной) регуляции. Примерами генов, продукты которых контролируют апоптоз клеток млекопитающих, являются bcl-2 (подавление), c-myc, p53 (усиление). Результат зависит от костимулирующих воздействий (например, от присутствия в среде цитокинов), типа клеток, структурных нюансов генных продуктов. Финал бывает неожиданным: вместо прогнозируемого апоптоза клетка вступает в митотический цикл, начиная пролиферативный процесс. Более того, некоторые из апоптозных генов относятся к группе протоонкогенов, т.е. генов, активирующих размножение клеток. Так, экспрессия c-myc в присутствии интерлейкина-3, эпидермального фактора роста или инсулиноподобного фактора-1 потенцирует пролиферацию клеток, тогда как без такого сопровождения активация с-myc запускает апоптоз. Повышение экспрессии нормального (дикого) р53 усиливает апоптоз, подавляя туморогенез; мутантный р53 лишается своей природной функции. До расшифровки этого факта р53 относили к онкогенам. Парадоксально, но решение идти по пути смерти или жизни часто принимается однотипными способами, и лишь совокупность определенных обстоятельств (определенных для клетки, но не ясных для наблюдателя!) влияет на окончательный результат.
Этот парадокс акцентирован в одной из крайних точек зрения: апоптоз является абортивным митозом, эволюция которого необратимо задерживается на одном из этапов — так называемых контрольных пунктах (англ. check points). Впрочем, такая диалектика сглаживается по мере углубления процесса, и с определенного этапа апоптоз располагает собственными эффекторами. Это соответствует тому, что постмитотические клетки, т.е. клетки, не способные к делению (например, нейтрофилы, нейроны, миоциты), сохраняют готовность к апоптозу.
На модели Drosophila melanogaster предложена гипотеза о центральном генезе (англ. reapergene) апоптозного процесса, который, обобщая информацию, идущую из разных каналов, формирует апоптозную программу. При этом допускается, что клетки постоянно борются за выживание, продуцируя антиапоптозные белки-протекторы. Их недостаток ведет к гибели, т.е. жизнь и смерть клетки воспринимаются как соревнование между белками-эффекторами, обладающими противоположными функциями. Его исход зависит от количественных соотношений, активности и стабильности апоптозных и антиапоптозных факторов, которые неодинаково выражены у разных клеток и весьма динамичны.
В этой связи принципиален вопрос — связан ли апоптозный процесс с новообразованием белка? Первое впечатление о том, что от этого зависит индукция апоптоза, оказалось неточным или, по крайней мере, не универсальным — апоптоз удается не только возбудить, но и ускорить при подавлении белкового синтеза. Это соответствует наблюдениям, согласно которым апоптозные изменения могут сдерживаться за счет перманентной продукции короткоживущих ингибиторов. Торможение синтеза белка блокирует этот механизм, возбуждая процесс. С этой точки зрения, понятны данные о том, что цитопласты (клетки, лишенные ядра и других органелл), сохраняют способность к апоптозу, впрочем, как и изолированные ядра, помещенные в бесклеточную среду. На этом основании можно говорить о том, что часть эффекторов апоптоза конституциональны, не требуют индуцированного синтеза белка. Отсюда изменения ядерного материала, о которых говорилось выше, следует признать вторичными, не имеющими пускового значения в апоптозном процессе. Очевидно также, что несмотря на сходный морфологический сценарий существуют различные по механизму варианты апоптоза, причем некоторые из них эпигенетичны.
В целом представление о генах-убийцах, привлекающее мистикой и элегантной простотой, служит лишь базой для современных концепций апоптоза, логично вписываясь в общую канву физиологии и патологии клеток. Не исключено, что специализированных апоптозных генов у высших организмов вообще не существует. В таком случае это обычные гены, задействованные в других физиологических реакциях, а их летальный эффект является результатом суперэкспрессии или коэкспрессии с другими генами. Вместе с тем базисная программа активной смерти клеток остается достаточно консервативной, сохранив единые принципы (даже структурное сходство генов) на разных уровнях эволюции, начиная от червей и до человека. Это позволяет экстраполировать и обобщать результаты, получаемые на разных экспериментальных моделях.
Эффекторы апоптоза
Любое активное изменение клеточного фенотипа опосредовано действием медиаторов, переводящих клетку в новое качественное состояние. Это может быть следствием растормаживания заблокированных генов либо активации/подавления преформированных молекул-эффекторов.
В реализации апоптоза принимает участие множество молекул, которые вступают в сетевые взаимодействия, преломляя активность первичных сигналов. Мы уже говорили об усилении активности Са2+/Mg2+-зависимой эндонуклеазы, с которой связана фрагментация ядерного хроматина, хотя это скорее символ, чем основополагающий механизм апоптоза. Обнаружена активация другого фермента, Са2+-зависимой трансглутаминазы-II, которая вызывает перекрестные сшивки, ведущие к необратимым структурным изменениям, таким как образование ригидной оболочки вокруг апоптозных тел и обнажение необычных для нормальной клетки мембранных компонентов, распознаваемых макрофагами по типу лектин-, фосфатидилсеринзависимых и иных взаимодействий.
Обращает на себя внимание кратковременный подъем Са2+ и циклического аденозинмонофосфата (цАМФ) в клетках, вступивших на путь апоптоза. Искусственное снижение или повышение уровня внутриклеточного Са2+ соответственно подавляет или индуцирует апоптоз. Складывается впечатление об апоптозных сигналах, исходящих от субкомпонентов мембранных фосфолипидов, прежде всего церамида, который образуется при индуцированном гидролизе сфингомиелина или заново синтезируется при активации специфических ферментов (церамидсинтетазы).
Центральную позицию занимают высокорестриктированные эндопептидазы, именуемые каспазами (англ. caspase, от cystein-aspartate-proteinase, т.е. цистеиновые протеиназы, расщепляющие пептидную связь вслед за аспартатом). Из дюжины известных каспаз по крайней мере восемь имеют отношение к апоптозу. Номера каспаз не соответствуют их порядку в апоптозном каскаде, и принятое деление на «инициаторы» и «исполнители» (англ. executioners) довольно расплывчато. Каспазы предсуществуют в цитозоле в виде неактивных предшественников (прокаспаз), но, подвергаясь ограниченному протеолизу (предыдущая каспаза расщепляет последующую), обретают эффекторную (протеолитическую) активность. Этому способствует и формирование надмолекулярных комплексов (апоптосом), которые образуются при гетеродимеризации каспаз с апоптогенными внутриклеточными белками или при гибридизации самих каспаз. Установлено, например, что каспаза-9 активирует каспазу-3 после связывания с апоптогенными факторами митохондрий (цитохромом с и Apaf-1) и АТФ, а каспаза-8 образует активные тетрамеры, принимая апоптогенную эстафету от адаптерных белков, ассоциированных с «доменами смерти» (англ. DD, death domain) Fas и TNFR1 (рецептор типа 1 для альфа-туморонекротического фактора). Это ведет к активации нижележащих эффекторов, которые, атакуя клеточные субстраты, напрямую включаются в развитие апоптозного фенотипа.
Мишенями для каспаз служат многочисленные белки, в том числе ферменты и ингибиторы, задействованные в клеточном гомеостазе. В целом участие каспаз в апоптозе образно сравнивают с хорошо продуманной и организованной военной операцией. Они отрезают гибнущую клетку, разрушая межклеточные контакты и ее внутреннюю инфраструктуру (цитоскелет). Расщеплению подвергаются гелсолин, ряд киназ, играющих важную роль в поддержании целостности цитоскелета, а также сами элементы цитоскелета, в частности актин. Каспазы участвуют и в разрушении командного центра клетки — ядра: прекращаются репликация и репарация ДНК, начинается ее фрагментация. Расщепляются также структурные компоненты ядерной оболочки, что ведет к дезорганизации и конденсации хроматина. Результаты этой разрушительной деятельности вынуждают клетку выбрасывать «белые флаги» — различные маркеры апоптоза, после чего вся кампания завершается фагоцитозом погибшей клетки.
Наряду с каспазами заметная роль в апоптозных реакциях принадлежит семейству Вcl-2-протеинов (от англ. B-cell lymphoma/leukemia-2). В различных типах клеток идентифицировано около полутора десятка аналогов Bcl-2, которые объединены в отдельное семейство благодаря наличию гомологичных доменов. Члены этого семейства различаются направлением действия на апоптоз и поэтому разделены на подсемейства — анти- и проапоптозное. Накопились данные о том, что гомологи Bcl-2 опосредуют свой эффект через митохондрии. Это особенно важно для реализации внутреннего (спонтанного) пути апоптоза, который определяет гибель клеток без внешних воздействий. Как оказалось, митохондрии формируют своеобразный стыковочный узел апоптозного процесса, обеспечивая взаимодействие между каспазами и представителями семейства Bcl-2 протеинов.
Проапоптозные гомологи Bcl-2, такие как Bax (от англ. Вcl-2 associated protein x) и Bak (от англ. Bcl-2 homologous antagonist/killer), нарушают целостность митохондриальной мембраны, формируя в ней поры и вызывая возникновение так называемой митохондриальной дисфункции. Это выражается в падении трансмембранного потенциала внутренней митохондриальной мембраны и выходе в цитозоль проапоптозных составляющих митохондрий (в частности, цитохрома с и Apaf-1), что сопровождается активацией каспазного каскада. В известной мере толчком к этому служит передислокация Вах из цитозоля в митохондрии, предшествующая активации каспаз.
Антиапоптозные аналоги Всl-2 выполняют противоположные функции. Они поддерживают целостность митохондриальных мембран, предотвращая перераспределение проапоптозных вариантов молекул. Отсюда следует, что изменение соотношения про- и антиапоптозных гомологов Bcl-2 играет важную роль в реализации апоптозного фенотипа клеток.
Несмотря на очевидную важность митохондрий в апоптозной программе, эти органеллы не являются абсолютно необходимыми для апоптоза. Об этом говорят наблюдения над гибелью цитопластов, которые представляют собой пузырьки цитоплазмы, лишенные ядер, гранул и митохондрий. Они подвергаются изменениям, напоминающим апоптоз интактных клеток, подтверждая предположение о том, что в цитозоле клеток содержится адекватный механизм для поддержания их запрограммированной гибели. Причины безмитохондриального апоптоза в точности не известны. Безусловную роль играют колебания тирозинфосфорилирования в сигналпередающих каскадах. Антиапоптозное действие ряда факторов сочетается с усилением тирозинфосфорилирования белков, а естественные (интерлейкин-10) или искусственные (генестин) ингибиторы тирозинкиназ отменяют задержку индуцированного (внешнего пути) апоптоза.
Судьба апоптозных клеток
Эволюционируя, апоптоз завершается вторичным некрозом, который можно обнаружить по повышению проницаемости плазматической мембраны для постмортальных красителей (трипановый синий, йодид пропидиума и др.). В отличие от первичного некроза, когда повреждение мембраны служит пусковым фактором клеточной гибели, при апоптозе это знаменует окончание процесса. Впрочем, в естественных условиях дело обычно не доходит до вторичного некроза, так как апоптотирующие клетки уничтожаются профессиональными (макрофаги) и непрофессиональными (например, фибробласты) фагоцитами. Макрофаги, поглотившие апоптозные клетки, обнаруживаются в очагах воспаления, а также в культурах in vitro.
Механизм распознания апоптозных клеток детерминирован необычными поверхностными структурами, которые появляются на внешнем слое плазматической мембраны и побуждают соседние клетки к фагоцитозу. Их образно называют сигналами «съешь меня» (англ. eat-me signals). Одним из кандидатов на роль такого рода сигнальных молекул служит фосфатидилсерин (ФС). В норме остатки ФС локализованы во внутреннем слое клеточной мембраны, но при апоптозе благодаря потере физиологической асимметрии мембранных фосфолипидов они выныривают на поверхность и распознаются специфическими рецепторами макрофагов. Существуют и более сложные пути, например кооперация между витронектиновым рецептором, коллагенсвязывающим белком CD36 и тромбоспондином. Полагают, что тромбоспондин, продуцируемый макрофагами, сопрягает их витронектиновые рецепторы и СD36 с неизвестным компонентом плазматической мембраны апоптозных клеток, инициируя поглощение стареющих клеток. Реакцию усиливает ряд цитокинов, которые действуют на ФС-зависимое поглощение апоптозных клеток. Поглощение заметно возрастает при адгезии макрофагов на внеклеточном матриксе, в частности на фибронектине. Моноциты не поглощают апоптозные клетки, но обретают такую способность после трансформации в макрофаги.
Недавно был описан новый механизм, позволяющий макрофагам осуществлять негативный отбор апоптозных клеток. Оказалось, что при встрече с макрофагами в живых клетках возникает отталкивающий сигнал, который прекращает это взаимодействие. Такую реакцию обеспечивает гомофильное взаимодействие CD31 на поверхности лейкоцитов и макрофагов. В апоптозных клетках передача сигнала нарушается, что ведет к более длительному контакту с макрофагами. Это облегчает узнавание вышеописанных маркеров на апоптозных клетках и их элиминацию.
Кроме сигналов «съешь меня» для привлечения фагоцитов апоптозные клетки секретируют особый хемотаксический фактор — фосфолипид лизофосфатидилхолин, для высвобождения которого требуется активность каспазы-3. Этот фактор стимулирует миграцию моноцитов и макрофагов в направлении его источника (апоптозных клеток), что обеспечивает своевременное и эффективное удаление погибающих клеток. Принципиально, что фагоцитоз апоптозных клеток не только не возбуждает секреции провоспалительных цитокинов, но даже подавляет ее благодаря аутокринному ингибированию. Не случайно своевременному удалению апоптозных клеток придается большое значение в предупреждении и обратном развитии воспалительных реакций.
Апоптоз и патология
Представления о запрограммированной смерти клеток, долгое время не привлекавшие внимания клиницистов, сегодня выдвинулись в число самых популярных и интригующих проблем медицины. Апоптоз ассоциируется с патогенезом злокачественных опухолей, атрофических (в частности, нейродегенеративных) и гиперпластических процессов, аутоиммунных заболеваний, вирусных инфекций. Каждая из приведенных ситуаций может быть проанализирована с позиций дисбаланса между нацеленностью клеток на выживание/пролиферацию и апоптоз. Направленная регуляция апоптоза просматривается как один из базисных принципов терапевтической стратегии будущего, и уже сегодня лечебный эффект многих цитотоксических противоопухолевых препаратов связывают с их способностью усиливать апоптоз. Перспективной выглядит идея регуляции экспрессии апоптозных и антиапоптозных генов при помощи антисмысловых олигонуклеотидов, блокирующих мРНК для определенных белков. Она уже получила подтверждение в опытах по торможению пролиферации лейкозных клеток при хронической миелоидной лейкемии.
Влияние на апоптоз (усиление или ослабление) оказывают различные микроорганизмы — вирусы, бактерии, грибы, простейшие. С этим связаны новые взгляды на патогенез инфекционной патологии. Примером могут служить представители семейства энтеробактерий: признаки апоптозной гибели обнаружены в культурах эпителиальных клеток и макрофагов, инфицированных шигеллами, сальмонеллами, эшерихиями, йерсиниями. Во многом это связано с межмолекулярными эффектами белков-токсинов, вводимых в клетку бактериями через аппарат секреции III типа. Они могут оказать влияние на клеточные гены в результате взаимодействия с протеинкиназами или иными факторами, контролирующими транскрипционный процесс. Не исключено, что данный механизм играет определенную роль в развитии энтеробактериальных инфекций (см. соответствующие разделы).
Установлено, что многие вирусы, вмешиваясь в регуляцию внутриклеточного гомеостаза, меняют соотношение между ростовыми и апоптозными потенциями инфицированных клеток. Известно более десятка вирусных генов, которые кодируют факторы, усиливающие апоптоз. Обнаружены они и у вирусов человека (аденовирусы, герпесвирусы, папилломавирусы, ортомиксовирусы, вирус иммунодефицита человека, вирус гепатита В и др.). В ряде случаев вирусиндуцированный апоптоз коррелирует с усилением экспрессии гена р53, претендующего на одну из ключевых позиций в индукции апоптоза. Такую гибель клеток удается предотвратить при помощи Bcl-2: выполняя функцию протоонкогена, он усиливает клеточную пролиферацию. Предложена гипотеза, согласно которой гибель CD4+ Т-лимфоцитов при ВИЧ-инфекции связана с апоптозом. Пусковым фактором служит взаимодействие наружного вирусного гликопротеина gp 120 с СD-рецепторами лимфоцитов. Этот сигнал запускает механизм, напоминающий апоптоз, инициируемый в системах Fas—FasL и ТНФ-a—ТНФR. В остальных случаях апоптоз возбуждается через проникновение вируса в клетку, требуя хотя бы частичной экспрессии его генома с образованием апоптогенных продуктов. Опережающая смерть клетки, несущей зачаток вируса — исход, выгодный скорее хозяину, чем вирусу. Не случайно об этом говорят как об альтруистическом самоубийстве, подразумевая, что клетка предпочитает погибнуть, исключив возможность размножения потенциально опасного вируса.
Логика подсказывает, что вирусы должны обладать и альтернативным механизмом, который, продлевая жизнь клеток, создавал бы условия для воссоздания как можно более многочисленного вирусного потомства в литическом цикле или обеспечивал переход вируса в латентное состояние (персистенцию). Действительно, некоторые вирусы синтезируют белковые продукты, которые блокируют апоптозный ответ инфицированных клеток. Это достигается разными способами, но их принцип сводится к двум механизмам — повышение экспрессии антиапоптозных (ростстимулирующих) генов клетки и инактивация эффекторных молекул апоптоза. К первой группе можно отнести стимуляцию протоонкогенов типа bcl-2 (это, в частности, наблюдается при персистенции вируса Эпстайна—Барр в В-лимфоцитах), ко второй — блокаду белка Р53, продукта апоптозстимулирующего гена р53 (например, Е6 — протеин вируса папилломы человека).
Известно, что вирусы вызывают и непрямой цитопатический эффект. В этом случае зараженные клетки атакуются эффекторами иммунитета, прежде всего CD8+ Т-лимфоцитами. В реализации цитотоксических потенций Т-лимфоцитов также участвует апоптоз, возбуждаемый в клетках-мишенях. В качестве вероятных механизмов рассматривается прямое действие эффекторных молекул, преформированных в Т-лимфоцитах, а также рецепторзависимая активация апоптозной программы атакуемых клеток. В первом случае действуют апоптозные протеазы (гранзимы), которые проникают в клетки-мишени через поры, образуемые в плазматической мембране перфоринами лимфоцитов. Во втором процесс запускается через системы Fas—FasL и ТНФ-a—ТНФR и является следствием активации собственного апоптозного аппарата клеток.
* * *
Концепция об апоптозе является одним из перспективных обобщений современной биологической науки. Сформулированная более трех десятилетий назад, она стала предметом всеобщего внимания после того, как было осознано, что клетки гибнут в результате активных реакций, поддающихся генетической и эпигенетической регуляции. Проблема оказалась сложнее, чем это выглядело после наблюдений на примитивных организмах. Вместе с тем именно первые факты, полученные в опытах на нематоде Caenorhabditiis elagans, стали основой для изучения клеточной смерти.
Будучи естественным механизмом структурного гомеостаза, апоптоз включается в патогенез многих болезней, в том числе инфекционной природы. С идеями апоптоза связано управление гиперпластическими, дегенеративными, атрофическими и воспалительными процессами, составляющими сущность онкологических, аутоиммунных, инфекционных и прочих заболеваний. Впереди много сюрпризов, которые не оставят равнодушными теоретиков и практиков медицины, послужив развитию представлений о живой материи и ее двух основополагающих процессах — жизни и смерти.
Механизмы противоинфекционного иммунитета
Все живое должно быть защищено.
А. Флеминг
- Ареактивные и реактивные механизмы противоинфекционной защиты.
- Колонизационная и антиинвазивная резистентность кожи и слизистых оболочек.
- Воспаление: мобилизация и функциональная кооперация эффекторов иммунитета.
- Клиренс крови: защита от внутрисосудистой инвазии.
- Иммунитет против внутримакрофагальных инфекций.
- Иммунитет против вирусов.
- Парадоксы иммунитета.
По степени опасности для человека микроорганизмы делятся на три категории — патогенные, условно-патогенные и непатогенные. Патогенные вызывают первичные инфекции, поражая здоровых людей. Условная болезнетворность отражает зависимость от дополнительных факторов и обстоятельств, которые в той или иной форме связаны с ослаблением иммунитета. Такие инфекции принято называть оппортунистическими, а их возбудителей — микробами-оппортунистами.
Оппортунистические инфекции — это прежде всего госпитальная патология, вторичные инфекции, или инфекционные осложнения, которыми так богата современная клиника, особенно хирургического профиля. Классической средой, где оживают микробы-оппортунисты, служат больные с врожденными или приобретенными дефектами иммунитета. Они поражаются вирусами, бактериями, грибами и простейшими, которые бессимптомно инфицируют здоровых людей и нередко входят в состав нормальной микрофлоры.
Непатогенные микроорганизмы безвредны для человека, хотя и могут быть опасны для других биосистем. Устойчивость к ним является атрибутом вида и наследуется подобно другим конституциональным признакам. Такой защиты может быть много для одних микробов и мало для других. С этим приходится мириться, так как изменить видовые признаки невозможно — их плюсы и минусы формировались тысячелетиями. «Крысы нечувствительны к дифтерийному токсину, и эта устойчивость наследуется так же, как крысиные уши и хвост».
О причинах невосприимчивости человека к микробным агентам, опасным для других животных, известно немного. Она может быть результатом неадекватности температурного режима, дефицита ростовых факторов или рецепторов для адгезии, подавления инвазивных и токсигенных функций. Например, развитие бруцелл в плаценте животных, приводящее к выкидышам, обеспечивается углеводом эритритолом. Он содержится в плацентах коров, овец, свиней, но отсутствует у человека, собак, крыс, кроликов, морских свинок. У них эта форма бруцеллеза не встречается. Неприживляемость или отсутствие условий для токсинообразования спасает от многих потенциально опасных микробов. Продукты некоторых бактерий очень токсичны, и можно отравиться (т.е. заболеть без инфекции), если они попадают в организм в готовом виде (например, при ботулизме). Животные не болеют дифтерией, но многие из них высокочувствительны к дифтерийному токсину.
Благодаря видовому иммунитету мы не замечаем множества микробов — они, по сути, выпадают из поля зрения медицинской микробиологии. Основное внимание сконцентрировано на патогенных и условно-патогенных агентах, иммунитет к которым относителен и во многом зависит от индивидуального опыта, т.е. приобретается или усиливается при контактах с возбудителем, точнее с его антигенами. Это вариант адаптации к потенциально опасным факторам внешней среды. Поэтому приобретенный иммунитет называют адаптивным, противопоставляя врожденной резистентности (англ. innate immunity), которая не усиливается после столкновений с инфекционными агентами и не оставляет после себя иммунологической памяти, т.е. способности к быстрому повышению резистентности при повторных встречах с тем же возбудителем. Врожденный иммунитет базируется на механизмах реактивной (индуцибельной) защиты, таких как фагоцитоз, активация комплемента, воспаление, секреторные реакции эпителиальных клеток (цитокины, катионные пептиды), синтез острофазных белков. Кроме того, устойчивость к инфекции зависит от механических, химических и биологических барьеров, конститутивно представленных в тканях, прежде всего на поверхности кожи и слизистых оболочек. Это факторы неспецифической резистентности в широком смысле.
Другими широкоизвестными противопоставлениями являются специфический (антигензависимый) и неспецифический (антигеннезависимый) иммунитет. Специфический и приобретенный иммунитет — синонимы, так как приобретение всегда есть результат высокоизбирательной (специфической) реакции на микробные антигены. Относительная устойчивость к инфекционным агентам базируется на факторах и механизмах неспецифической резистентности, но обретает завершенность благодаря эффекторам специфического иммунитета.
Колонизационная резистентность кожи и слизистых оболочек
Чтобы спровоцировать болезнь, возбудитель обязан решить по меньшей мере две задачи — закрепиться и размножиться в зоне заражения. Это называется колонизацией, и от того, насколько быстро она произойдет, зависит судьба инфекта, т.е. его превосходство в конкуренции противоборствующих сторон. Помешать колонизации — самый простой и надежный способ заблокировать развитие инфекционного процесса. Многие, по выражению И.И. Мечникова, «вредные деятели» терпят здесь неудачу, оказываясь бессильными против факторов колонизационной резистентности. Это особенно вероятно, если инфицирующая доза невелика. Используя высокие дозировки «человеческих микробов», удается заразить животных, которые в естественных условиях к ним невосприимчивы.
Неповрежденная кожа непроницаема для большинства инфекционных агентов, а секретируемые ею продукты (молочная кислота, жирные кислоты, хлористый натрий, катионные пептиды) действуют биоцидно и/или биостатично. Это обеспечивает способность кожи к самоочищению — показатель, который не без основания предложено считать индикатором здоровья. Если на кожу нанести бактериальную культуру, то спустя полчаса или меньше число живых бактерий резко уменьшится. В той же культуре, взятой из стеклянной чашки или с кожи трупа, бактерии сохраняют жизнеспособность. Вот почему для прохождения сквозь кожу микробы используют повреждения эпидермиса или прибегают к помощи кровососущих насекомых. Надо обладать уникальными способностями, чтобы самостоятельно преодолеть такого рода барьер. Из бактерий, пожалуй, только Staphylococcus aureus и Propionobacterium acne достаточно экипированы для этого, хотя и здесь обычно прослеживается связь с микротравмами.
В связи с высокой резистентностью кожи большинство инфекций начинается со слизистых оболочек. Они выстилают пищеварительный канал, мочеполовые органы, дыхательный тракт, т.е. те пути, по которым идет основной поток веществ из окружающей среды. Все, что усиливает сопротивляемость слизистых оболочек, повышает устойчивость к инфекции, и, наоборот, любые нарушения нормального состояния слизистого (мукозального) тракта снижают этот показатель. Классическая концепция о местном иммунитете во многом опирается на представления о колонизационной резистентности слизистых оболочек.
Поверхность слизистых оболочек непрерывно омывается секретами. Они обладают выраженной антиколонизационной активностью, так как содержат антиадгезивные, биостатические и биоцидные факторы. Жидкость перемещается в одном направлении благодаря мукоцилиарному транспорту (реснитчатый эпителий), перистальтике гладкой мускулатуры, гидростатическому давлению. Этим, а также непрерывным обновлением (слущиванием) эпителиальных клеток достигается механическое очищение слизистых оболочек от случайных микробов. Препятствием для колонизации является и нормальная микрофлора, прежде всего бактерии, которые в изобилии заселяют кишечник, ротовую полость, верхние отделы респираторного тракта, влагалище. Их значение определяется не только конкуренцией за сайты адгезии, но и формированием вокруг себя среды, негативно действующей на чужие (эктопичные) микробы. Бактерии-резиденты выделяют антибиотические вещества, меняют содержание кислорода и рН, поддерживают физиологическое воспаление в слизистой оболочке и регулируют иммунные реакции, влияя на функциональную кооперацию иммунокомпетентных клеток. Отсюда понятно, почему состояние нормальной микрофлоры вызывает повышенный интерес, побуждая оберегать ее от нарушений, например, при лечении антибиотиками.
Антиколонизационные факторы неодинаковы и порой уникальны для разных отделов слизистой оболочки. Это справедливо для биоцидности кислой среды желудка, антибактериальной активности желчи и пищеварительных соков тонкого кишечника, мощной системы макрофагов, фиксированных в альвеолах и бронхах, колонизационного барьера, создаваемого анаэробной флорой толстого кишечника, гликогензависимого закисления среды молочнокислыми бактериями влагалища и др.
Нельзя не удивляться эффективности, с которой работают механизмы, обеспечивающие стерильность здоровых легких, мочевого пузыря, полости матки. Легкие содержат комплекс белков, которые входят в состав сурфактанта и не только выполняют важные функции, сопряженные с дыханием, но и помогают уничтожать бактерии, действуя как опсонины. Это объясняется тем, что сурфактантные белки относятся к семейству коллектинов, т.е. содержат лектиновый центр (он связывается с углеводными радикалами бактерий) и коллагеноподобный фрагмент, рецептируемый макрофагами. Кроме того, сурфактантные белки усиливают опсонические эффекты комплемента и антител. К сожалению, они бессильны против капсульных бактерий (в частности, пневмококка): капсула экранирует углеводы их клеточной стенки.
Базисная антиколонизационная активность секретов усиливается за счет антител, продуцируемых лимфоидной тканью слизистых оболочек. Они относятся к иммуноглобулинам класса A, которые транспортируются на поверхность слизистых оболочек при помощи секреторного компонента — особого белка, синтезируемого эпителиальными клетками (рис. 1). Некоторые бактерии (например, менингококки и гонококки) продуцируют специфические IgA-протеазы. Разрушая IgA-антитела, они лишают их главной функции — способности подавлять адгезию инфекционных агентов на эпителиальных клетках.

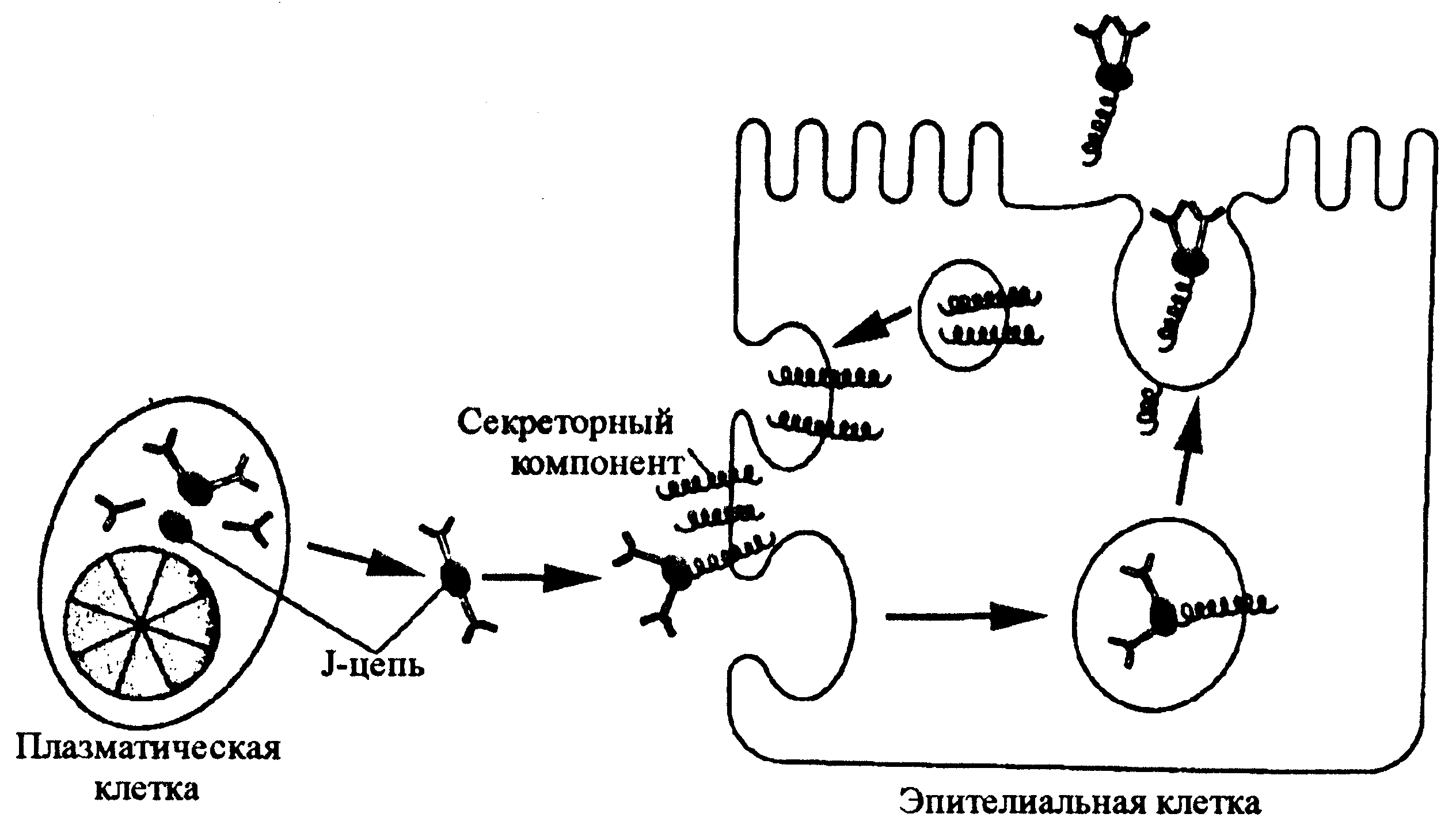
Лимфоидная ткань, ассоциированная со слизистыми оболочками, работает в собственном режиме и в известном смысле автономна от системного иммунитета. Ее лимфоциты, стимулированные антигенами и настроенные (по выражению иммунологов, коммитированные) на синтез IgA-антител, даже после выхода в циркуляцию обычно возвращаются «домой», в субэпителиальную ткань слизистых оболочек, предпочитая решать местные задачи. Это так называемый хоминг-эффект (англ. homing — стремление к дому, очагу), который объясняется сродством лимфоцитов к рецепторам (адрессинам) эндотелия посткапиллярных венул, дренирующих мукозальный тракт (рис. 2). На избирательности хоминг-эффекта основана энтеральная вакцинация, нацеленная на создание иммунитета в масштабе общей системы слизистых оболочек путем перорального введения микробных антигенов.

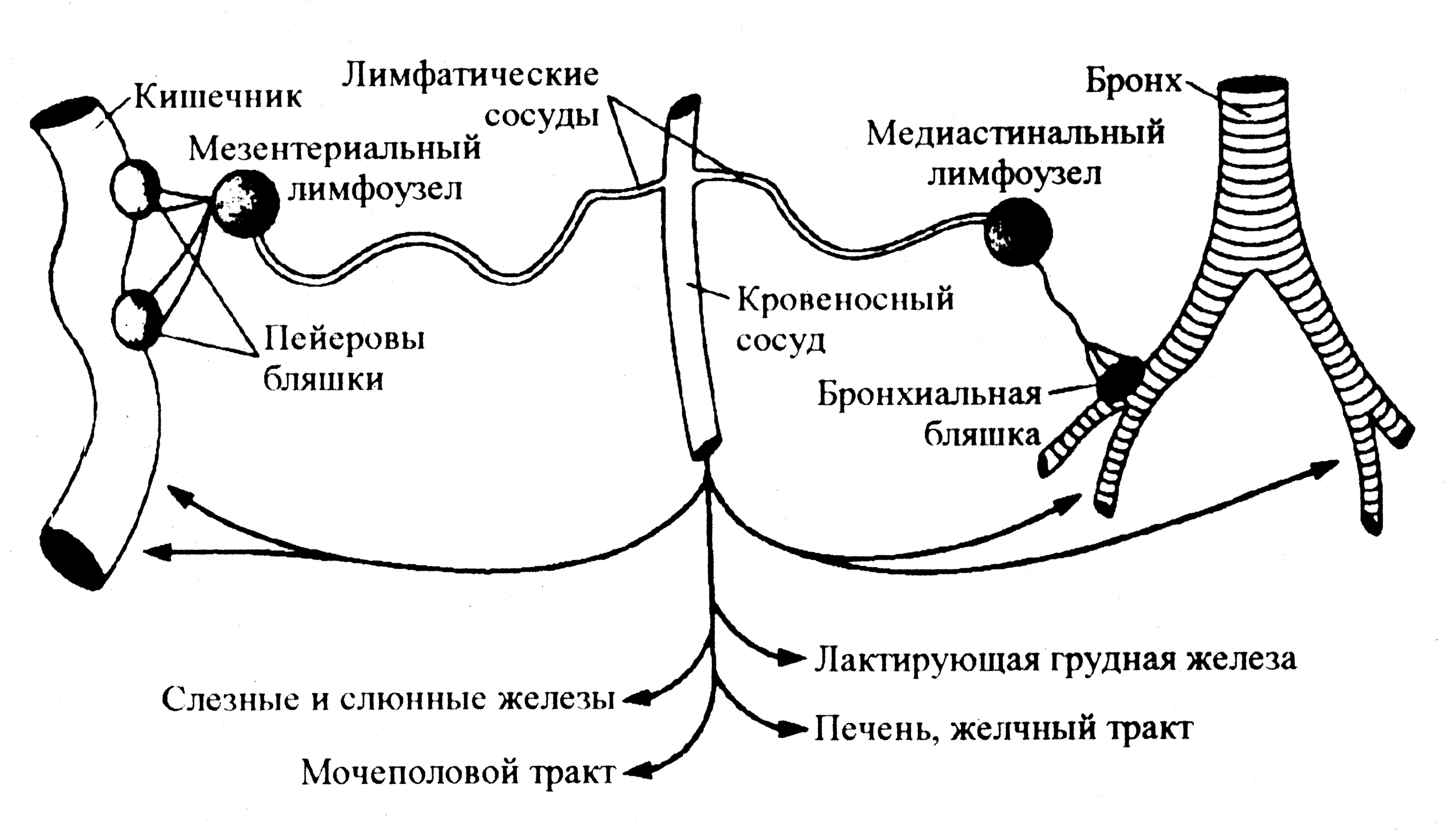
Следует отметить еще один важный момент. Гомеостаз мукозального тракта во многом зависит от непрерывной регенерации эпителиоцитов, которая обеспечивается пролиферацией и дифференцировкой стволовых клеток, встроенных в слизистую оболочку. В кишечнике эту функцию выполняют клетки крипт, откуда дифференцированные энтероциты мигрируют в ворсинки и, отработав, сбрасываются в полость кишечника.
В криптах непосредственно под стволовыми клетками располагается особый тип клеток (клетки Панета), которые секретируют антибактериальные факторы — лизоцим и катионные пептиды (криптидины), похожие на дифенсины фагоцитов. Они защищают стволовые клетки от микробной контаминации, поддерживая обновление энтероцитов и сохранение целостности эпителиального покрова. О тяжелых последствиях нарушения регенерации слизистой оболочки свидетельствует картина поражения кишечника при лучевой болезни и цитостатической терапии. Из-за гибели быстро делящихся стволовых клеток и вторичного повреждения эпителиального барьера (клинически это проявляется кровоизлияниями в полость кишечника) заметно возрастает опасность септических инфекций, исходящих из кишечного тракта.
Иммунитет и воспаление
Поверхность кожи и слизистых оболочек — первый рубеж, где оказывается сопротивление инфекционным агентам. Защита здесь носит бесконфликтный характер и в основном определяется факторами, которые имеются до заражения и не требуют дополнительных реакций для своего образования или активации (исключение — антигениндуцированный синтез секреторных антител и продукция эпителиальными клетками цитокинов и катионных пептидов).
Картина меняется после проникновения (инвазии) микробов в субэпителиальную ткань. Это всегда вызывает реакцию, так как инвазии сопутствует повреждение. Стандартным ответом является воспаление, которое либо обрывает конфликт без серьезных последствий, либо разрастается до крупных осложнений. Диалектика воспаления сурова — пожертвовать частью ради целого, т.е. организма. Не дифференцируя ткани по жизненной важности, воспалительный процесс развивается везде, где нарушен гомеостаз и налицо виновник повреждения. Так, в частности, возникают абсцессы легкого, печени, мозга и т.д. Воспаление травмирует ткани, сокращает полезный объем органа и служит источником токсических начал, отравляющих организм.
Мысль об иммунитетной функции воспаления утвердилась как одно из следствий фагоцитарной теории И.И. Мечникова. Логика проста: если фагоциты защищают от микробной инвазии, то и реакция, которая обеспечивает их мобилизацию в зону поражения, выполняет ту же функцию. Безусловно, учение о воспалении шире, чем учение об антимикробной защите, но именно здесь гомеостатический смысл этого механизма проявляется особенно ярко.
Реакцию начинают «клетки тревоги» (тучные клетки и макрофаги рыхлой соединительной ткани), которые первыми реагируют на повреждение, извещая об опасности и необходимости продолжения реакции. Они секретируют множество медиаторов, которые вместе с производными плазмы повышают проницаемость сосудов, вызывая эмиграцию лейкоцитов из сосудистого русла. Это означает, что воспалительная реакция развивается благодаря последовательному подключению гуморальных факторов и клеток — резидентных (постоянно присутствующих в соединительной ткани) и мобилизуемых из крови. Данный принцип базируется на сложном (сетевом) переплетении эффекторных и регуляторных молекул, которые обслуживают воспалительный процесс, обрывая или способствуя его развитию.
Известны различные варианты воспаления. Вместе с тем, опираясь на природу его главных эффекторов (фагоцитов), можно говорить о двух основных разновидностях — нейтрофил- и макрофагзависимом воспалении. Первое носит гнойный характер и вызывается гноеродными (пиогенными) бактериями. Нейтрофилы (их изобилие придает воспалительному экссудату гнойный характер) охотно принимают помощь гуморальных факторов — комплемента и антител, которые в очаге воспаления действуют как опсонины, усиливая фагоцитарные реакции. Нейтрофилзависимое воспаление отличается острым экссудативно-деструктивным течением, но иногда обретает вялотекущий, хронический характер, являясь основой хронической пиогенной патологии. Макрофаги служат эффекторами гранулематозного воспаления, которое исходно протекает хронически с преобладанием пролиферативно-склерозирующего компонента и усиливается Тh1-цитокинами. Такие реакции наблюдаются при заражении туберкулезной палочкой, листериями, возбудителями проказы, бруцеллеза, токсоплазмоза, лейшманиоза и др. Их объединяет способность к длительному выживанию (персистенции) внутри макрофагов (см. ниже).
Защита от внутрисосудистой инвазии
Чтобы попасть в кровь, инфекционный агент должен преодолеть барьер из лимфатических узлов, располагающихся по ходу лимфатических сосудов. Здесь много макрофагов, которые, занимая стратегически выгодную позицию в сосудистых синусах, извлекают из лимфы чужеродных агентов. Впрочем, и сама тканевая жидкость не безобидна для микробов: в лимфе содержатся комплемент, антитела и другие элементы плазменного транссудата, которые обладают прямой антимикробной активностью или усиливают реакции макрофагов, выступая в роли опсонинов.
Резидентные факторы лимфы и лимфатических узлов не всегда способны уничтожить возбудителя. В таких случаях здесь, как и в субэпителиальной ткани, возникает воспаление (лимфаденит), причем основной удар принимают близлежащие (регионарные) узлы. Воспалительный процесс (об этом говорит увеличение и болезненность лимфоузлов) задерживает распространение инфекции не только за счет уничтожения микробов, но и благодаря снижению скорости лимфотока, который при сильном разбухании и повреждении лимфоузла может вообще прекратиться. Но и это не всегда останавливает инфекцию. Возбудитель проникает в эфферентную лимфу и далее в кровоток. Иногда инвазия в лимфу и кровь происходит бессимптомно. К примеру, некоторые вирусы и другие внутриклеточные паразиты проникают в кровяное русло, инфицируя (но не повреждая) моноциты или лимфоциты, которые разносят их по организму еще в инкубационном, т.е. доклиническом периоде.
Уцелеть в кровеносном русле рядовым микробам тоже непросто. Прежде всего сама кровь — не очень подходящая среда для размножения бактерий. Лишь после прогревания или разведения она становится пригодной для этих целей. Кровь — продукт многих тканей, и поэтому в ней содержится множество биологически активных начал, включая антимикробные (биоцидные, биостатические, опсонические) факторы. В виде резидентных (лимфа) или мобилизуемых при воспалении факторов их действие продолжается в тканях, где оседают микробы.
Но обезвреживание микроорганизмов, попавших в кровь, происходит не только в циркуляции. Они удаляются макрофагами ретикулоэндотелиальной системы, главным образом печени, легких, селезенки и костного мозга. Макрофаги, встроенные в сосудистые синусы, подобно щупальцам, захватывают проплывающие мимо них чужеродные частицы и молекулярные агрегаты, особенно те, которые покрыты опсонинами плазмы — антителами, производными комплемента, фибронектином и другими белками (например, С-реактивный белок; липополисахаридсвязывающий фактор; коллектины). Если резидентные макрофаги не справляются с задачей, на помощь приходят фагоциты крови (нейтрофилы, моноциты) — возникает очаг воспаления. Это объясняет, почему при септических метастазах в реакцию прежде всего вовлекаются ткани, богатые резидентными макрофагами.
Системные реакции на микробную инвазию
Фагоциты, сосредоточенные в очаге воспаления, ведут борьбу не только в непосредственной близости от возбудителя. При активации они выделяют цитокины, которые, поступая в кровь, оказывают гормоноподобное действие на различные клетки. Именно под влиянием цитокинов развиваются симптомы неспецифической интоксикации, общие для всех инфекций. Как и местное воспаление, это адаптивная реакция, которая становится опасной лишь при переходе определенных границ.
Наиболее показательна температурная реакция — лихорадка. Она возникает при действии на терморегуляторные центры гипоталамуса интерлейкинов-1, -6, фактора некроза опухолей-a (ФНО-a) и ряда других цитокинов, объединяемых в понятие «эндогенные пирогены». Микробы, поражающие теплокровных животных и человека, наиболее активны при 36—37°С, т.е. при нормальной температуре тела. Повышение температуры ослабляет их жизнедеятельность, в том числе болезнетворность. Поэтому температура служит одним из факторов видового иммунитета, сортирующего микробы на тех, против которых мы обязаны реагировать, и всех остальных, которых мы просто не замечаем. Птицы не чувствительны к бациллам сибирской язвы, но если температуру цыпленка (41—42°С) понизить до 37°С, заражение ведет к патологии. Этот знаменитый опыт Л. Пастера, повторенный многократно на других животных с другими возбудителями, наглядно свидетельствует о значении температурного фактора для антиинфекционной резистентности.
Цитокины активированных макрофагов принадлежат к ключевым медиаторам так называемого острофазного ответа, главными и универсальными индукторами которого являются воспаление и травма. Реакция острой фазы сопровождается значительными изменениями сывороточной концентрации ряда факторов, известных как острофазные белки (ОФБ). Они синтезируются гепатоцитами и в меньшем количестве другими клетками, прежде всего макрофагами и фибробластами.
Белки, содержание которых при острофазной реакции возрастает (иногда в сотни и тысячи раз), называются позитивными ОФБ. К ним относятся два главных ОФБ — С-реактивный белок и амилоид А, а также фибриноген, гаптоглобин, a1-антихемотрипсин, a1-антитрипсин, факторы В и С3 системы комплемента, эндотоксин/липополисахаридсвязывающий белок, некоторые коллектины. Представителями негативных ОФБ (их концентрация в плазме снижается при острофазном ответе) являются альбумин и трансферрин.
По крайней мере семь цитокинов способны непосредственно индуцировать синтез ОФБ гепатоцитами. Лидерами являются интерлейкин-1, -6 и ФНО-a. Их опережающим источником служат активированные макрофаги. Острофазный ответ усиливается глюкокортикоидами, которые повышают число высокоаффинных цитокиновых рецепторов на гепатоцитах. В свою очередь, синтез глюкокортикоидов потенцируется теми же цитокинами, формируя одно из звеньев системной реакции на микробную инвазию и интоксикацию.
Хотя значение острофазной реакции до конца не изучено, не вызывает сомнений, что она играет гомеостатическую роль. ОФБ действуют как ингибиторы протеиназ, опсонины, факторы свертывания и репарации тканей. Повышение скорости оседания эритроцитов (индикатор активности воспалительных процессов) отражает изменение сывороточной концентрации ОФБ, прежде всего фибриногена.
Реакции адаптивного (специфического) иммунитета
Иммунология возникла как прикладная наука. Она родилась из стремления управлять устойчивостью к болезням, искусственно воспроизводя то, что создала природа — высокоизбирательную антимикробную резистентность, приобретаемую после латентных или клинически значимых контактов с инфекционными агентами. Селективность базируется на клоноспецифическом распознавании антигенов В- и Т-лимфоцитами, которое лежит в основе сложных реакций, возбуждаемых антигенными стимулами в иммунной системе. Их результатом является образование антител и Т-эффекторов, нацеленных на элиминацию антигенов. Об этом мы говорили в специальном издании, поэтому здесь будем предельно кратки.
При общей стратегии антитела и Т-лимфоциты (т.е. эффекторы гуморального и клеточного иммунитета) воспринимают разную информацию об антигенах, отличаясь по функциям в противоинфекционном иммунитете. Антитела реагируют со свободными антигенами, что на иммунологическом языке означает «антигены, не ассоциированные с молекулами главного комплекса гистосовместимости (МНС)». Это могут быть целые микробы, молекулы или субмолекулярные фрагменты. В отличие от антител, Т-эффекторы (CD4 и CD8) распознают антигенные пептиды в сочетании с МНС (у человека они называются HLA — от англ. human leukocyte antigens). Мишенью для них служат зараженные клетки, которые, переработав микробные антигены, выносят их фрагменты на свою поверхность в комплексе с МНС/HLA.
Реактивность к антигенам обеспечивает пластичность противоинфекционного иммунитета и возможность его направленной регуляции. Если неспецифическая резистентность является эволюционным завоеванием вида и изменить ее в порядке личной инициативы невозможно, то специфический иммунитет растет вместе с хозяином. Он отражает его иммунологический опыт (т.е. историю встреч и борьбы с антигенными раздражителями), а потому всегда индивидуален. Каждый новорожденный начинает свой путь практически с нуля.
Это нельзя понимать буквально. Способность приобретать иммунитет — наследуемый признак, так как лимфоциты даются с рождения. Но они служат лишь реквизитом для образования антител, Т-эффекторов и клеток памяти, т.е. всего того, чем определяется полноценность антимикробной защиты и управление ее базисными (врожденными) ресурсами.
Бессмысленно противопоставлять друг другу различные факторы и механизмы иммунитета, выискивая их преимущества и недостатки — они эффективны лишь в содружестве. При первом контакте с антигеном специфические реакции развиваются медленно, в течение нескольких суток, что является минусом, допуская возможность опережающего размножения возбудителей. Фагоциты и комплемент намного оперативнее, особенно с учетом резидентного пула макрофагов соединительной и ретикулоэндотелиальной тканей. Впрочем, и их подключения может не потребоваться на фоне надежного колонизационного барьера эпителиальных покровов. Нередко этого достаточно, чтобы снять угрозу или по крайней мере выиграть время для адаптивной перестройки иммунитета. Иными словами речь идет об эшелонированности антимикробной защиты (см. таблицу).
Иммунитет против внутримакрофагальных инфекций
За исключением риккетсий и хламидий бактерии способны размножаться вне живых клеток. Многие из них сохраняют эту тенденцию в инфицированном организме и, проникая в ткани, реплицируются внеклеточно. В основе связанной с ними патологии лежит гнойное воспаление, поэтому такие бактерии называются пиогенными (от лат. pyos — гной). Главным эффектором специфического иммунитета в таких случаях являются антитела. Они нейтрализуют микробные токсины и, выступая в роли опсонинов, усиливают реакции нейтрофилов и комплемента. Этот тройственный союз (нейтрофилы—комплемент—антитела) определяет практически всю устойчивость к пиогенным инвазиям.
От классических пиогенных агентов мало чем отличаются возбудители, которые, проникнув в клетки, не задерживаются здесь надолго. Это характерно для гонококков, шигелл, йерсиний и ряда других бактерий. Они инвазируют быстро обновляющиеся эпителиоциты слизистых оболочек и к тому же ускоряют их гибель своими токсинами. В защите против таких бактерий ведущая роль тоже принадлежит антитоксическим и антиинвазивным антителам, действующим в содружестве с нейтрофилами и комплементом.
Факторы и механизмы противоинфекционного иммунитета (принцип эшелонированности антимикробной защиты)
| Уровень действия | Механизмы |
| Эпителиальные покровы: кожа | Механический барьер Механическое самоочищение: шелушение Химическое самоочищение: жирные кислоты (секрет сальных желез), молочная кислота, NaCl (пот), катионные пептиды |
| слизистые оболочки | Антиадгезивные факторы: IgA-антитела, продукты секретов Механическое самоочищение: вымывание, мукоцилиарный транспорт, перисталь-тика, чихание, кашель, отслойка поверхностных пластов эпителия Биоцидные и биостатические факторы секретов: лизоцим, пероксидаза, лактоферрин, катионные пептиды, кислая реакция (желудочный сок, влагалищный секрет), желчные кислоты Макрофаги, встроенные в эпителий Нормальная микрофлора |
| Субэпителиальная ткань | Резидентные факторы: тканевая жидкость (см. «Кровь»), клетки (тучные клетки, макрофаги/гистиоциты) Мобилизуемые факторы: воспалительная реакция |
| Лимфоидный барьер | Резидентные факторы: лимфа (см. «Кровь»), макрофаги лимфатических узлов Мобилизуемые факторы: воспалительная реакция |
| Кровь | Факторы плазмы: комплемент, антитела, белки острой фазы, бета-лизины, лизоцим, коллектины и др. Макрофаги ретикулоэндотелиальной системы (печень, селезенка, легкие, костный мозг) |
| Внутренние органы | См. «Субэпителиальная ткань» |
Радикального своеобразия можно ожидать от инфекций, возбудители которых длительно персистируют в клетках, не повреждая их (по крайней мере структурно), и благодаря этому изолируют себя от антител и эффекторов острого воспаления. Клетки, способные обеспечить такое убежище, должны отвечать, как минимум, трем условиям: 1) активно фагоцитировать, 2) отличаться доступностью и мобильностью и 3) быть долгожителями. Эти признаки лучше всего сочетаются у макрофагов, которые присутствуют во всех тканях, подвижны, являются профессиональными фагоцитами и способны к длительному (месяцы) выживанию. Не случайно некоторые бактерии, грибы и простейшие научились использовать макрофаги как среду обитания. Длительное выживание внутри макрофагов связано с тем, что такие микробы умеют обезопасить себя от биоагрессивного потенциала фагоцитов. Они препятствуют образованию биоцидной ячейки — фаголизосомы (блокада слияния фагосом и лизосом), ускользают из фагосомы в цитоплазму, инактивируют биотоксичные молекулы или попросту резистентны к ним.
Таких агентов немного, но они играют большую роль в патологии человека (туберкулез, проказа, бруцеллез, листериоз, токсоплазмоз, актиномикоз и др.). В основе лежит гранулематозное воспаление, которое развивается в ответ на медленное, но перманентное повреждение макрофагов. Процесс поддерживают моноциты, мобилизуемые из крови, а в роли катализатора выступают сенсибилизированные T-хелперы (точнее один из их субвариантов — Th1), которые активируют макрофаги, пробуждая их от функциональной депрессии.
Последовательность событий сводится к следующему. Микробные антигены экспрессируются на поверхности зараженных макрофагов в комплексе с молекулами главного комплекса гистосовместимости и стимулируют Т-хелперы, вызывая секрецию цитокинов, усиливающих агрессивность макрофагов (главная роль отводится гамма-интерферону). Это способствует уничтожению внутриклеточных паразитов, а также активирует соседние (еще не зараженные) клетки, настраивая их на уничтожение не только микробов, возбудивших реакцию, но и других, попавшихся под руку агентов. Иммунологи называют такие макрофаги вооруженными, или рассерженными, подразумевая их повышенную агрессивность. Антитела тоже образуются, но это антитела-свидетели. Они не играют существенной роли в иммунитете, так как внутриклеточные мишени для них недоступны.
Т-зависимые реакции макрофагов не всегда спасают от утвердившейся инфекции, которая, продолжая эволюционировать, ведет к разрастанию и распаду гранулем. Иными словами, действуя без разбора, активированные макрофаги могут наносить вред собственным тканям, расширяя зону повреждения. Но это уже неизбежная диалектика воспаления и иммунитета, которая отражает неадекватность антигенной нагрузки, являясь основой иммунопатогенеза.
Иммунитет против вирусов
Вирусы являются облигатными внутриклеточными паразитами, т.е. размножаются только в живых клетках. Проникая в чувствительные (пермиссивные) клетки, они распадаются на субкомпоненты, высвобождая свою геномную молекулу (ДНК или РНК), которая, проходя через этапы репликации, транскрипции и трансляции, обеспечивает копирование вирусных генов и белков. Из них собираются новые вирусные частицы (вирионы), способные заражать соседние клетки, запуская очередные циклы вирусного онтогенеза. Этот вариант, называемый репликативной, или продуктивной инфекцией, часто ведет к гибели зараженных клеток и клинически воспринимается как острая патология. Альтернативой служит стабилизация внутриклеточного вируса с развитием персистентной инфекции. Она лежит в основе хронической вирусной патологии и ее обострений.
В связи со сказанным следует различать две главные мишени для антивирусных реакций: 1) свободные вирионы, попадающие в организм при первичном заражении или высвобождающиеся из клеток при продуктивной инфекции и 2) инфицированные клетки, экспрессирующие на своей поверхности вирусные антигены.
Вирионы атакуются антителами, протективность которых определяется блокадой вирусных рецепторов. Превентивный эффект антител хорошо выражен при инфекциях с длительным инкубационным периодом, когда вирус перед тем, как достичь своих мишеней, должен пройти через циркуляцию (т.е. через этап вирусемии), где он нейтрализуется даже небольшим количеством антител. Фаза вирусемии обязательна для натуральной и ветряной оспы, вирусов гепатита, кори, полиомиелита и др. Профилактическая вакцинация дает здесь надежный эффект, а перенесенная инфекция оставляет стойкий гуморальный иммунитет.
Если репликативная инфекция все же состоялась (внедрившийся вирус не был нейтрализован) и процесс набрал деструктивную силу, его развитие может быть задержано путем блокады новых вирионов, которые высвобождаются из клеток. Антитела способствуют решению этой задачи и, если иммунный ответ не слишком запаздывает, помогают остановить заболевание, действуя как механизм выздоровления. Иначе обстоит дело с инфекциями, при которых поражаемый орган служит одновременно входными воротами для вируса. Такие заболевания (например, грипп и другие острые респираторные инфекции) имеют короткий инкубационный период, и образование сывороточных антител не поспевает за их развитием. Основное значение имеют местные (секреторные) IgA-антитела.
Но, как уже говорилось, не все вирусы ведут себя столь прямолинейно. Вместо быстрого уничтожения зараженных клеток, они длительно используют их как экологическую нишу или, по крайней мере, задерживают цитолиз или апоптоз. К примеру, оболочечные вирусы, играющие едва ли не главную роль в патологии человека, распространяются на соседние клетки путем вирусиндуцированного слияния клеточных мембран с образованием импластов. При этом сам вирус не покидает зараженных клеток, оставаясь неуязвимым для антител. Еще более коварна вирогения, когда вирус сохраняется в клетке в виде геномной молекулы — свободной (неинтегративная вирогения) или встроенной в хромосомы хозяина (интегративная вирогения). Это создает предпосылки для вялотекущей патологии с периодическими обострениями. Антитела не обеспечивают удаления таких вирусов из организма: главную задачу решают Т-лимфоциты.
В отличие от антител единственной мишенью для Т-лимфоцитов служат зараженные клетки. Инфицированная клетка (если находящийся в ней вирус хотя бы частично экспрессирует свои гены) содержит вирусные пептиды, которые появляются на клеточной поверхности в виде коинтегратов с молекулами МНС. Именно они (комплексы МНС— вирусные пептиды) служат объектом распознавания и атаки для Т-лимфоцитов. Не случайно сегодня большое внимание привлекает создание вакцин для направленной стимуляции Т-клеточного иммунитета. Их, в частности, планируется использовать для элиминации персистентных вирусов.
Антивирусная активность Т-лимфоцитов определяется тремя механизмами:
- уничтожением вирусинфицированных клеток (это закреплено в понятии цитотоксичность);
- элиминацией внутриклеточного вируса;
- использованием цитотоксического и антивирусного потенциала других клеток-эффекторов.
В первом случае лимфоциты вызывают цитолиз, повреждая плазматическую мембрану, но чаще используют иной, более изящный способ — индуцируют апоптоз, растормаживая внутреннюю программу смерти клеток-мишеней (см. «Болезнетворность вирусов»).
Гибель собственных клеток — не идеальный вариант защиты. Если вирус обладает слабой цитотоксичностью (а тем более лишен ее), хозяин больше страдает не от самого вируса, а от последствий деструктивных реакций, связанных с агрессивностью клеток-эффекторов. Понятие иммунопатогенез достаточно популярно в вирусологии и используется применительно к ряду острых и хронических инфекций (инфекционный мононуклеоз, вирусные гепатиты, лимфоцитарный хориоменингит и др.). Элиминация внутриклеточного вируса без повреждения клетки выглядит более рационально. В принципе это возможно, так как активированные Т-лимфоциты секретируют g-интерферон, который тормозит вирусную репликацию. Однако его продукция часто запаздывает, начинаясь лишь тогда, когда зараженные клетки уже несут на себе (экспрессируют) вирусные антигены, воспринимаемые Т-клетками. Кроме того, в отличие от a- и b-интерферонов (см. ниже) g-интерферон обладает слабым антивирусным эффектом, в большей степени опираясь на помощь активируемых им макрофагов и NK-клеток (естественных киллеров).
Что касается опосредованных эффектов, то они связаны с тем, что зона, где совершаются реакции Т-лимфоцитов, не остается без внимания других клеток-эффекторов, прежде всего макрофагов. Последние (точнее моноциты, которые после выхода из крови трансформируются в макрофаги) привлекаются сюда Th1-цитокинами и, подвергаясь активации, включаются в борьбу с вирусом. Макрофаги не только усиливают гибель инфицированных клеток, но, продуцируя собственные интерфероны (a, b), способствуют излечению от вируса зараженных клеток и повышают устойчивость их соседей.
Кроме антигенных пептидов, распознаваемых Т-лимфоцитами в комплексе с молекулами МНС, нелитические (оболочечные) вирусы экспрессируют на клеточной мембране белки своего суперкапсида. С иммунологической точки зрения, это свободные антигены, так как они не ассоциированы с молекулами главного комплекса гистосовместимости. Такие антигены распознаются В-лимфоцитами и, соответственно, антителами, давая повод для размышлений о значении антител в уничтожении вирусинфицированных клеток. Действительно, связываясь с вирусными антигенами на поверхности клеток, антитела опсонизируют их, делая мишенью для фагоцитов, естественных киллеров и комплемента. Но легко демонстрируемый в модельных опытах, этот механизм (антителозависимая клеточная цитотоксичность) остается в тени Т-зависимых реакций против вирусинфицированных клеток. Более того, антитела могут экранировать антигены от Т-лимфоцитов, снижая эффективность антивирусной атаки.
Разговор об устойчивости к вирусам мы начали с антигензависимого (специфического) иммунитета, для развития которого требуется довольно много времени. Между тем так же, как при любой инфекции, вирусы получают отпор с момента инфицирования. Это достигается благодаря факторам, которые предсуществуют до заражения или активируются тотчас после него. Среди последних лучше всего изучены интерфероны — разновидность цитокинов с антивирусной активностью. Существует три типа интерферонов (a, b и g), которые различаются по происхождению, физико-химическим свойствам и биологической активности. Интерфероны a и b (интерфероны типа I) секретируются всеми клетками, хотя и сохраняют свои исторические названия — лейкоцитарный (a) и фибробластный (b). Интерферон g (интерферон типа II) продуцируется главным образом активированными Т-лимфоцитами и естественными киллерами и поэтому называется иммунным. Известно более 20 разновидностей g -интерферонов и по одной a- и b-интерферонов.
Впервые о вирусной интерференции заговорили в 1957 г., когда было обнаружено, что вирусинфицированные клетки секретируют фактор, подавляющий репликацию вируса в соседних клетках. Механизм этого явления сводится к следующему. При заражении вирусом клетка начинает секретировать интерфероны, которые связываются со специфическими рецепторами соседних клеток. a- и b-интерфероны реагируют с общим рецептором, для g-интерферона имеется отдельный рецептор. Связывание интерферонов побуждает клетку к синтезу по крайней мере двух ферментов, которые блокируют синтез вирусных белков, разрушая вирусные мРНК и подавляя их трансляцию на рибосомах (рис. 3). Благодаря торможению вирусной репликации создается барьер из клеток, устойчивых к вирусу и сдерживающих его распространение. То, что это действительно важно, доказывают опыты на животных: блокада интерферонов антителами в сотни раз повышает чувствительность мышей к вирусной инфекции. Кстати, для интерферонов характерна видовая специфичность: каждый вид животных имеет собственные интерфероны и не чувствителен к чужим аналогам.
Интерфероны a и b образуются быстро (в течение 24 ч) и поэтому служат важным фактором экспресс-защиты; g-интерферон секретируется гораздо позднее (после дифференцировки Т-эффекторов) и сам по себе слабо влияет на репликацию вирусов. Антивирусный эффект g-интерферона скорее определяется тем, что он повышает МНС-II-зависимую экспрессию вирусных антигенов на поверхности зараженных клеток, делая их более заметной мишенью для Т-хелперов, а также усиливает цитотоксические функции макрофагов и естественных киллеров. Действие a- и b-интерферонов тоже не ограничено подавлением внутриклеточного вируса. Они стимулируют экспрессию молекул МНС-I, повышая эффективность цитотоксических реакций CD8 Т-лимфоцитов.

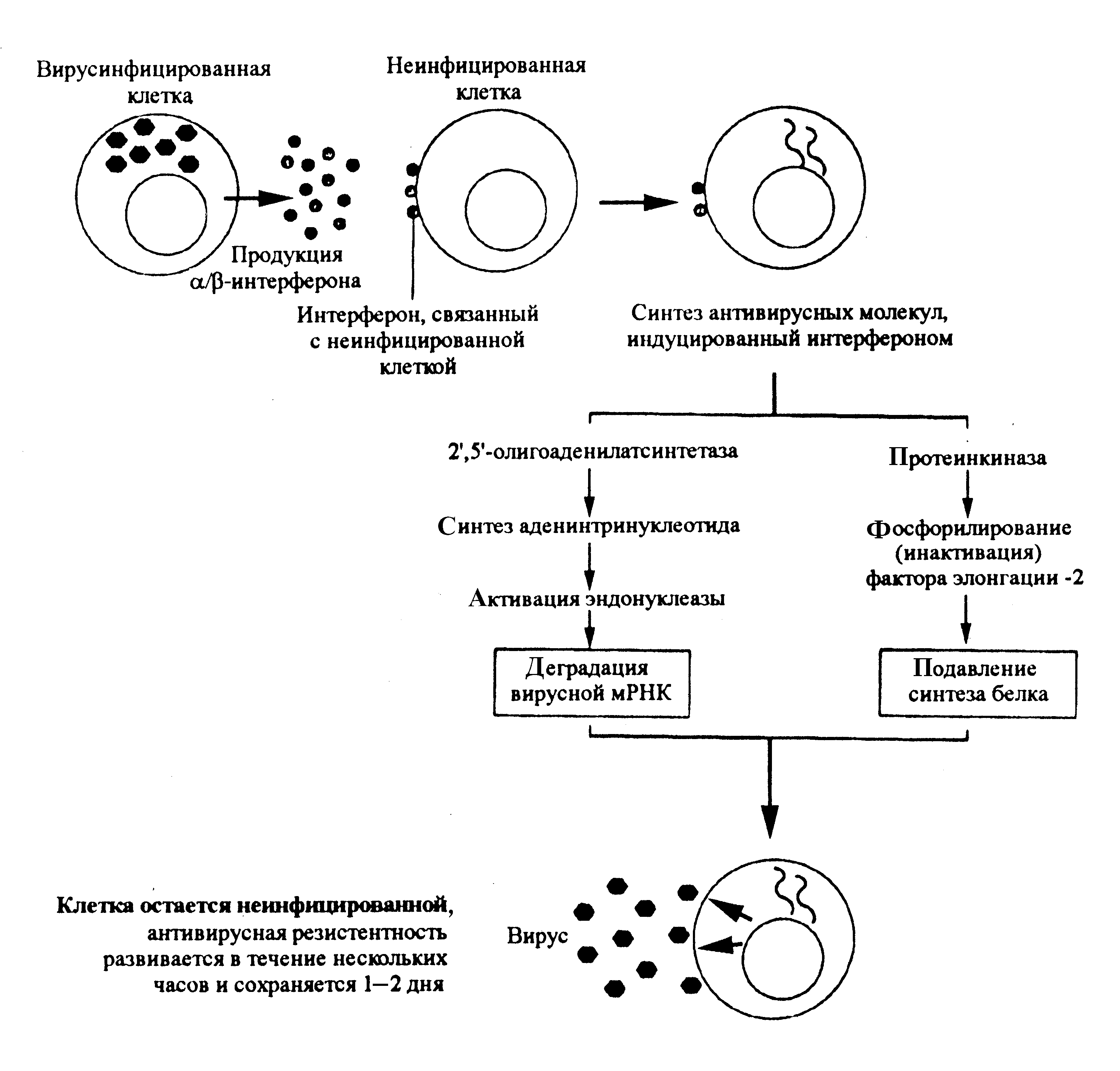
Кроме вирусов, интерфероны усиливают резистентность к риккетсиям, микобактериям, простейшим. Это неудивительно, так как, подобно вирусам, эти микробы размножаются внутри клеток, делая их мишенью для Т-лимфоцитов и антирепликативного эффекта интерферонов. Способностью индуцировать синтез интерферонов (речь идет об a- и b-интерферонах) обладают многие микробные продукты (например, липополисахаридные эндотоксины) и синтетические вещества (полианионы, двухспиральные РНК). Они называются интерфероногенами и так же, как сами интерфероны, используются в клинике.
После того, как интерфероны научились получать генно-инженерным способом, рынок постоянно пополняется рекомбинантными интерферонами, которые испытываются при различных вирусных и невирусных инфекциях. Контролируемые опыты говорят об эффективности a-интерферона при хронических гепатитах В и С, герпетических и ряде других инфекций. Интерфероны применяются и в лечении опухолей, особенно при злокачественных заболеваниях крови. Расчет строится на их универсальной антипролиферативной активности, которая проявляется в торможении синтеза вирусных, но и (хотя более слабо) клеточных белков. Имеет значение и то, что интерфероны усиливают эффекторные функции макрофагов и естественных киллеров, а также МНС-зависимую презентацию антигенов Т-лимфоцитам.
К сожалению, клинические результаты, как правило, хуже того, что можно было ожидать, исходя из высокой протективной активности естественно образующихся интерферонов. Кроме того, подобно другим цитокинам, интерфероны имеют множество мишеней в организме и при длительном применении дают осложнения, препятствующие продолжению терапии (лихорадка, слабость, мышечные боли, токсичность для почек, печени, костного мозга, миокарда и пр.).
Кроме интерферонов в доиммунном (т.е. до приобретения специфического иммунитета) надзоре за вирусинфицированными клетками принимают участие естественные киллеры (NK-клетки). Это неоднородная популяция клеток, которые морфологически похожи на лимфоциты или несколько крупнее (большие гранулярные лимфоциты). NK лишены рецепторов, распознающих антигены, и потому не способны к специфическим (антигензависимым) реакциям. В них есть гранулы, которые содержат цитотоксические факторы, аналогичные перфорину, гранзимам и гранулолизину Т-киллеров. В периферическом пуле лимфоцитов NK представлены неравномерно. В крови они составляют 10—15% мононуклеаров, доминируя среди так называемых О-клеток (лимфоциты, лишенные маркеров В- и Т-лимфоцитов). Их много в печени, селезенке и костном мозге, мало в лимфатических узлах, слизистых оболочках и практически нет в тимусе.
Особенностью NK-клеток является двойственность механизма распознавания клеток-мишеней. Они располагают двумя типами рецепторов: одни активируют, а другие блокируют NK-цитотоксичность. Активирующие рецепторы распознают структурную чужеродность клеток, блокирующие — взаимодействуют с молекулами МНС-I. Связывание с МНС-I инициирует сигнал, подавляющий NK-агрессивность. Это защищает нормальные ткани от NK-повреждения, предрасполагая к уничтожению клеток, дефектных по МНС-I. Такие клетки возникают при вирусных инфекциях и опухолевых процессах. Они неуязвимы для Т-киллеров, и именно здесь NK-реакции представляются наиболее значимыми.
NK убивают вирусинфицированные и опухолевые клетки. Подобно макрофагам, они могут делать это без посторонней помощи, но такие реакции лишь частично раскрывают их эффекторные ресурсы. Активация Th1-цитокинами (прежде всего ИЛ-2) значительно усиливает агрессивность NK-клеток и расширяет спектр атакуемых объектов.
Кроме цитотоксичности NK-клетки наделены медиаторными функциями. При активации они секретируют цитокины (g-интерферон, ФНО-a, ИЛ-8, ГМ-КСФ и др.), которые в зависимости от обстоятельств оказывают стимулирующее или супрессорное воздействие на лимфоциты, моноциты/макрофаги, кроветворные клетки и пр. В этом отношении они близки другим клеткам иммунной системы, таким как Т-лимфоциты, макрофаги и тучные клетки.
Проблема противовирусного иммунитета получила отражение и в концепции апоптоза в связи с представлениями о так называемом альтруистическом самоубийстве клеток. Подразумевается, что, реагируя на вирусную инвазию, клетки предпочитают погибнуть, чтобы опередить репликацию вируса, опасного для своих соседей и организма в целом. Некоторые вирусы препятствуют этому: блокируя апоптоз, они создают условия для продуктивной инфекции. Это достигается разными способами, но принцип сводится к двум механизмам — повышение экспрессии антиапоптозных (ростстимулирующих) генов клетки и инактивация эффекторных молекул апоптоза. Такую функцию несут гены, экспрессируемые в начале репликативного цикла. Поздние вирусные гены могут кодировать факторы, которые усиливают апоптозный процесс, содействуя цитопатогенному эффекту.
***
Вряд ли будет ошибкой, если тактическое и стратегическое разнообразие антимикробной защиты поставить в эволюционную зависимость от патогенетической изощренности инфекционных агентов. Всякое действие рождает противодействие, и именно многоликость микробных инвазий и интоксикаций определила потребность в усложнении механизмов иммунитета. Несмотря на впечатляющий арсенал конститутивных и индуцибельных механизмов, аппарат противоинфекционной защиты лишен совершенства. Впрочем, он и не может быть безупречен, так как ему противостоит эволюция инфекционных агентов, стремящихся к общей для всего живого цели — выжить и закрепиться в биосфере. По адаптации к новым условиям микробы значительно превосходят высшие организмы. В известной мере это компенсируется системой иммунного реагирования, функционирующей благодаря совместным усилиям факторов врожденного и адаптивного иммунитета.
Симбиоз держится не только на согласии, но и на противоречиях. Они могут решаться путем компромисса или открытых конфликтов. Экологическая система «макро-микроорганизм», которой, с одной стороны, управляет симбионт (в более узком смысле — паразит), а с другой — иммунитетные силы хозяина, изобилует такой диалектикой. Именно в их противоборстве рождаются болезни и одновременно выздоровление, т.е. то, что именуется инфекционным процессом.
Принципы диагностики инфекционных заболеваний
Нонешние годы мудрены народы.
- Чувствительность и специфичность диагностики: ложноположительные и ложноотрицательные результаты.
- Микроскопия исследуемого материала.
- Культуральный метод диагностики.
- Выявление микробных антигенов и специфических метаболитов.
- Тесты, основанные на определении нуклеиновых кислот.
- Выявление антител и сдвигов в системе клеточного иммунитета.
- Достоинства и недостатки разных способов диагностики.
История диагностики того или иного заболевания отражает эволюцию наших представлений о нем. Это великолепно подходит к способам диагностики инфекционных болезней, которые в своем развитии прошли дистанцию от микроскопии исследуемых образцов до современного молекулярного анализа. В основе усовершенствований лежит стремление повысить качество метода, увеличив его точность, чувствительность и простоту исполнения. Эти, казалось бы, несовместимые требования явились фундаментом, на котором формировались классические и открывались новые подходы к выявлению инфектопатологии.
Оценка любого из методов предполагает сравнение частоты истинно положительных (совпадающих с присутствием возбудителя в материале) и истинно отрицательных (совпадающих с отсутствием патогенного микроба) результатов. Это достигается использованием двух показателей — чувствительности и специфичности. С учетом ложноположительных (истинно позитивных) и ложноотрицательных (истинно негативных) результатов они высчитываются следующим образом: 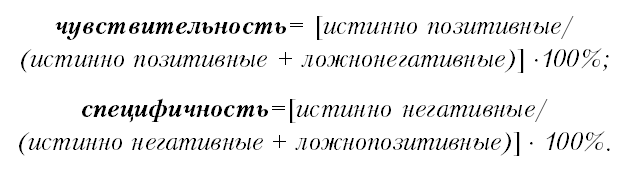

Микроскопия
Микроскопическое обследование материала было первым анализом, который применяли для подтверждения инфекционной природы заболевания. Так были открыты возбудители амебной дизентерии, парши, кандидоза, гонореи, туберкулеза, хламидиоза, риккетсиозов и многих других протозойных, фунгальных и бактерийных инфекций. Позже, с появлением электронного микроскопа, принцип микроскопии стали использовать для обнаружения вирусов и доказательства их участия в патогенезе инфекционного процесса.
Но оказалось, что далеко не все, что приносит пользу в раскрытии природы заболевания, обладает чувствительностью и специфичностью, необходимыми для диагностического исследования. Действительно, если почти все гельминтные, протозойные и фунгальные инвазии сегодня выявляются путем прямой микроскопии клинического материла, морфология большинства бактерий слишком проста для такой идентификации. Впрочем, есть несколько примеров, когда это возможно. Во-первых, диагностика сифилиса. Здесь учитывается характерная форма и подвижность спирохет в свежевзятых или фиксированных (окрашенных серебром) мазках из первичного и вторичных поражений. Другим примером служит определение бактерий в жидкостях, которые в норме стерильны. Так диагностируют пиогенный менингит, гонорею, эмпиему плевры. Наконец, следует отметить выявление микобактериальных инфекций, например открытых форм туберкулеза. В этом случае проводят анализ мокроты на присутствие кислотоустойчивых бактерий. Их в норме нет в респираторном тракте, и поэтому наличие таких бактерий говорит о легочном туберкулезе.
Хотя вирусы не видны под световым микроскопом, вирусиндуцированные изменения клеток могут иметь диагностическое значение. Примером являются многоядерные симпласты в мазках из поражений при простом герпесе или опоясывающем лишае, а также специфические внутриклеточные включения в тканях, инфицированных цитомегаловирусом или вирусом бешенства.
Повышение специфичности микроскопии достигается путем использования меченых (конъюгация с флюорохромом) антител против подозреваемого патогена (прямой метод; рис. 1) или против антител, которые применялись на первой стадии анализа (непрямой метод; рис. 2).

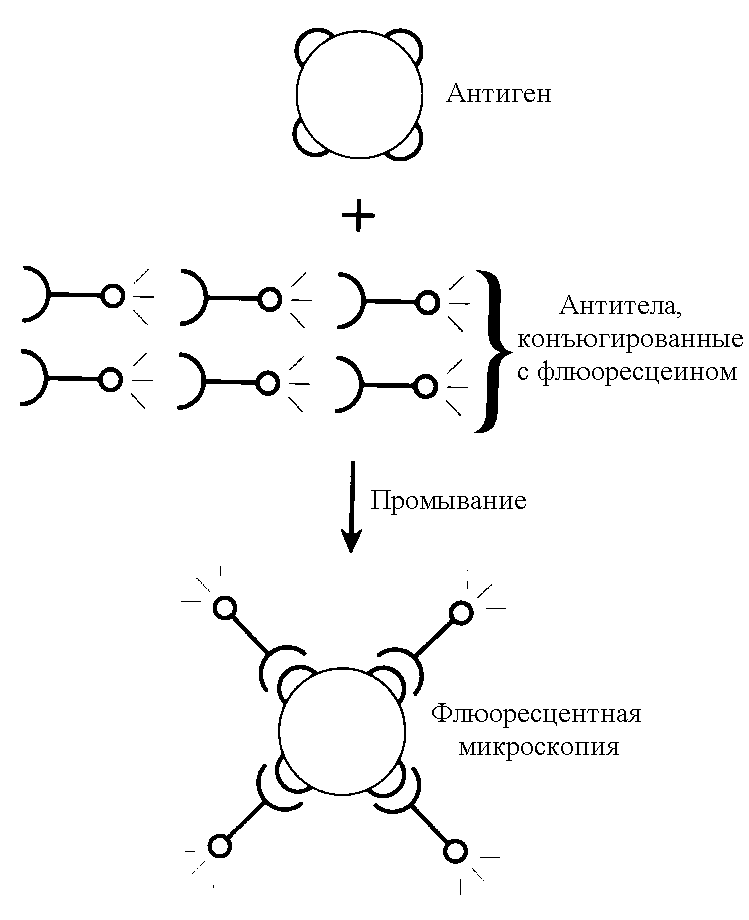

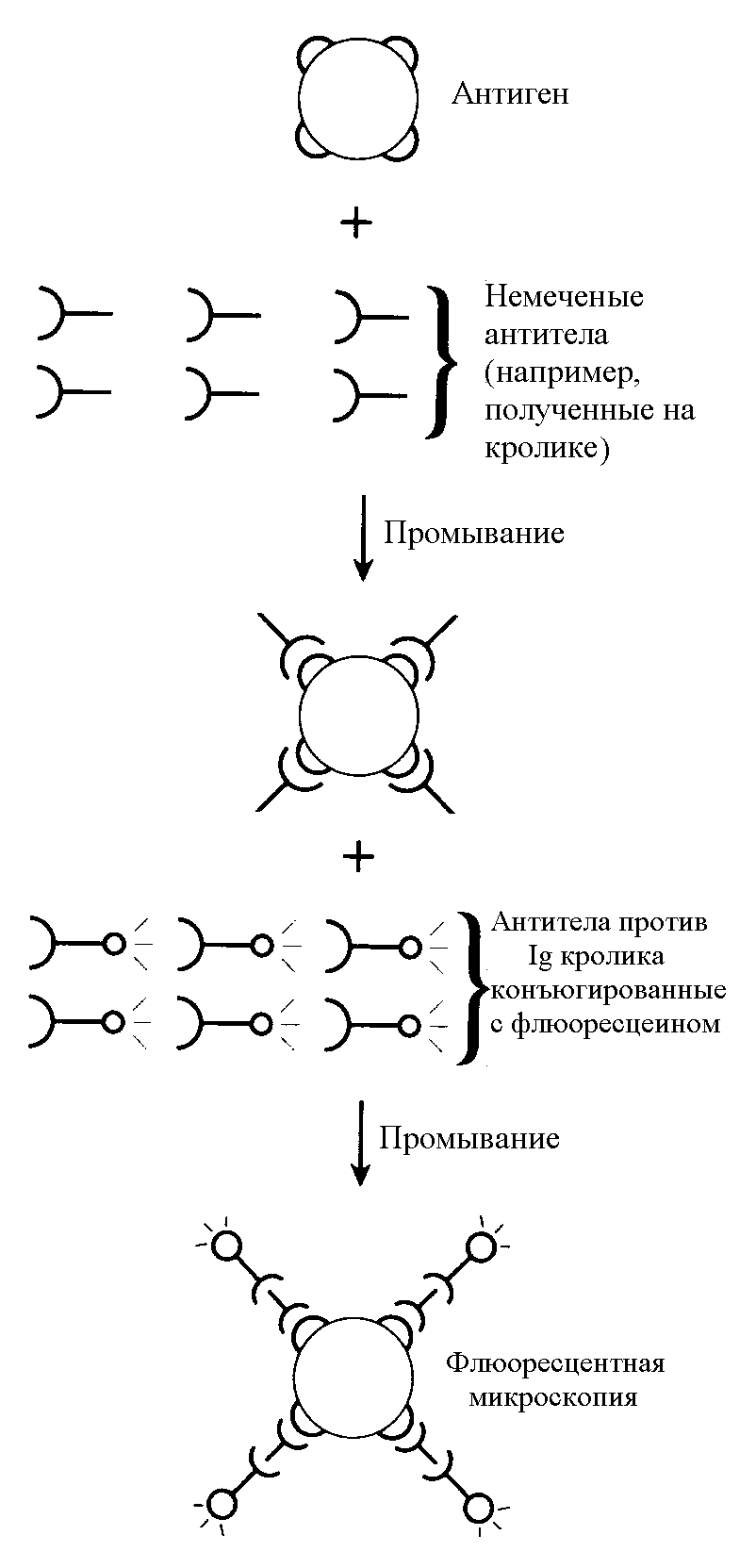
После отмывания препарат анализируют под флюоресцентным микроскопом. Метод может применяться для индикации любых патогенов, против которых получены антитела. Лучше использовать моноклональные антитела, каждое из которых нацелено против единственного антигенного эпитопа и поэтому в отличие от обычных (поликлональных) антител не будет давать нежелательных перекрестных реакций с другими микроорганизмами. Это обстоятельство справедливо для всех тестов, основанных на выявлении антигенов. К сожалению, иммунофлюоресцентная микроскопия с моноклональными антителами вследствие разных причин не получила широкого распространения.
Культуральный метод
Принцип метода заключается в идентификации возбудителя на основе выделения чистой культуры. Идентификацию проводят при помощи микробиологических, биохимических, иммунохимических и генетических маркеров. Изолированные штаммы можно хранить, готовить вакцины, изучать чувствительность к лекарственным препаратам, механизмы вирулентности, анализировать эпидемиологические закономерности и т.д. Возникло специальное направление, занятое разработкой и производством питательных сред, своего рода поваренной книги микробиологии. Современные прописи рекомендуют среды разного назначения, которые позволяют выделять нужную культуру из нестерильных участков тела (селективные среды, подавляющие постороннюю микрофлору), дифференцировать возбудителя по виду колоний и ферментативным признакам (дифференциальные среды) или комбинировать вместе и то и другое.
Анализ культур стал рутинным методом диагностики многих бактерийных и фунгальных инфекций. Отрицательный результат не означает возможности присутствия возбудителя в исследуемом материале и, наоборот, выделение некоторых микробов-оппортунистов не говорит об их обязательном участии в заболевании у данного больного. Это связано с тем, что чувствительность культурального метода зависит от техники взятия и хранения материала, количества микроба в исследуемом объекте, качества питательной среды и условий культивирования. Надо заметить, что анализ (получение чистой культуры бактерий) занимает длительное время и в лучшем случае продолжается не менее 24—48 ч. К тому же известно, что некоторые бактерии и грибы не культивируются in vitro или их культивирование сопряжено с нерентабельными затратами. По этой же причине культуральный метод не используется для выявления гельминтных и протозойных агентов. Трудности возникают с вирусами, хламидиями и риккетсиями, которые требуют для выделения специальных клеточных культур.
Выявление микробных макромолекул
Антигены. Мысль о том, что о присутствии микробов в исследуемом материале можно судить по молекулам, специфичным для возбудителя, возникла вместе с представлениями об антигенном устройстве живых организмов. Серологическое (иммунохимическое) выявление антигенов остается одним из основных способов микробиологической индикации.
Чувствительность диагностики определяется разрешающей способностью иммунохимического метода. Методики, основанные на внешних эффектах (агглютинация, преципитация, связывание комплемента), требуют для своего воспроизведения значительных количеств антигена и антител и поэтому лишены необходимой чувствительности. Тем не менее, они используются для выявления ряда инфекций, например в диагностике бактериального (менингококк, пневмококк, палочка инфлюэнцы, тип b) или фунгального (Cryptococcus neoformans) менингитов. В этих случаях применяется агглютинация латексных частиц, покрытых специфическими антителами против капсульных полисахаридов.
Современные тесты позволяют фиксировать наличие антигена без вторичных проявлений иммунохимических реакций. Это методы первого уровня, обладающие максимальной чувствительностью. Сюда относятся иммунофлюоресцентный, радиоиммунный и иммуноферментный (иммуноэнзимный) анализы. Наиболее широко используется твердофазный вариант иммуноферментного анализа, в котором антигены вылавливаются антителами, фиксированными на полистироловом планшете, а затем проявляются (в прямой или непрямой аранжировке) мечеными антителами (рис. 3).

Специфичность иммунохимического анализа повышает применение моноклональных антител на каждом из этапов реакции. Как уже говорилось, это повышает избирательность взаимодействия антител с антигеном, так как моноклональные антитела связываются с единственным эпитопом.
Специфические микробные метаболиты. Фенотипическое своеобразие микробов проявляется и в особенностях их токсического и ферментативного профиля. Поэтому определение данных показателей в исследуемом материале может оказаться полезным в диагностике инфекционного процесса. Примером служит выявление Helicobacter pylori у больных с пептическими язвами желудка и 12-перстной кишки. Дело в том, что H. pylori, обитающий в слизистой оболочке желудка, выделяет мощную уреазу — фермент, который, превращая мочевину в аммиак и углекислый газ, нейтрализует окружающую кислую среду. Определение уреазы является оптимальным методом диагностики хеликобактерной инфекции, так как содержимое желудка не содержит других бактерий, обладающих уреазной активностью. Для этого можно обследовать биопсийный материал, но эндоскопия является небезопасной процедурой и к тому же (если материал получен неадекватно) может дать ложноотрицательный результат. Этих недостатков лишен респираторный тест с мочевиной. Обследуемый принимает с пищей мочевину, меченную радиоактивным углеродом. Под влиянием уреазы из нее выделяется углекислый газ, который после всасывания в кровь выделяется легкими. Его регистрируют в выдыхаемом воздухе, начиная с первых минут после приема мочевины. Метод высоконадежен, так как отражает суммарную метаболическую активность бактерий независимо от места колонизации.
Нуклеиновые кислоты. Развитие молекулярной генетики позволило установить еще один класс взаимокомплементарных молекул — нуклеиновые кислоты, считываемые в клетке в процессе репликации и транскрипции. Стремление к повышению качества диагностических манипуляций инициировало принципиально новый подход к индикации микроорганизмов. Он базируется на обнаружении нуклеиновых кислот, точнее их фрагментов, специфичных для каждого вида микроорганизмов. Фактически речь идет об идентификации генов, своеобразие которых определяется последовательностью четырех пурин-пиримидиновых оснований, детерминирующих различные фенотипические признаки.
Все тесты, основанные на манипуляциях с нуклеиновыми кислотами, можно разделить на две группы: выявление генов при помощи фрагментов ДНК (РНК), добавляемых к исследуемому материалу, и варианты цепной реакции, предназначенные для амплификации генов, т.е. для их искусственной репликации in vitro.
Гибридизация. Двойная спираль ДНК построена из дискретных нитей, удерживаемых вместе водородными связями между комплементарными основаниями (аденин—пурин, гуанин—цитозин). Водородные связи отличаются непрочностью, поэтому двухцепочечная ДНК может быть разделена на одиночные нити при нагревании до 95—100оС. Если температуру понизить, то в результате случайных молекулярных взаимодействий комплементарные нити реассоциируют, образуя двухспиральную молекулу. Это происходит только в тех случаях, когда обе нити представляют гомологичную (генетически родственную) пару. Из каждой нити ДНК можно выбрать и искусственно синтезировать короткий фрагмент (зонд), который будет взаимодействовать только с комплементарной для себя последовательностью ДНК-мишени. Это служит основой для высокоспецифичных гибридизационных тестов, предназначенных для строго избирательного выявления молекул ДНК по наличию в них последовательностей, гомологичных известным ДНК-зондам.
Так как последовательности, из которых складываются дискретные гены, кодируют все структурные и функциональные признаки микроорганизмов, всегда удается отыскать (а затем синтезировать) уникальные последовательности нуклеотидов, специфичные для определенной группы микробов. Более того, гибридизационные тесты не ограничены спариванием ДНК-ДНК; стабильные гибриды образуются и в системах ДНК-РНК и РНК-РНК. Например, для гибридизации часто используется 16S рибосомальная РНК, которая по последовательности нуклеотидов отличается у разных видов и представлена тысячами идентичных копий в каждой клетке в отличие от фенотипически значимых генов, для большинства которых имеется единственная копия.
Технически гибридизационные тесты сводятся к следующему. Одноцепочечный ДНК-зонд метится изотопом, ферментом или любой другой легко распознаваемой меткой. Исследуемый материал обрабатывают в режиме, позволяющем разрушить микроорганизмы, высвободить и денатурировать ДНК, т.е. расчленить ее на одиночные нити. После инкубации зонда с исследуемым образцом измеряют количество меченой ДНК, связавшейся (вступившей в гибридизацию) с нуклеиновыми кислотами тест-препарата. Обычно перед добавлением меченого ДНК-зонда нуклеиновые кислоты исследуемого материала фиксируют на твердофазных сорбентах. Это позволяет легко удалить несвязанные меченые зонды путем отмывания. Но реакцию можно проводить и в растворе. В этом случае образовавшиеся комплексы ДНК (зонд—мишень) избирательно фиксируются на специальных сорбентах после завершения гибридизации.
Для получения положительного результата исследуемые образцы должны содержать несколько тысяч микробов. Чувствительность повышается после подращивания в культуре. Например, некоторые ДНК-вирусы удается идентифицировать в тканевых культурах до проявления цитопатического эффекта. Точно также ДНК-гибридизация существенно ускоряет обнаружение микобактерий, для получения видимых колоний которых требуется потратить 4—6 нед, а затем еще несколько дней на идентификацию. Объективности ради заметим, что в подобных случаях хорошо срабатывают и серологические тесты, выявляющие микробные антигены.
Полимеразная цепная реакция (ПЦР). Чувствительность микробиологического анализа, основанного на выявлении нуклеиновых кислот, можно многократно (теоретически неограниченно) повысить путем их амплификации, т.е. мультикопирования специфических фрагментов ДНК in vitro. Речь пойдет о классическом варианте цепной амплификации, который широко используется в современной диагностике инфекционных заболеваний.
Чтобы спланировать ПЦР-тест, необходимо знать строение искомой нуклеиновой кислоты, т.е. последовательность нуклеотидов того участка, который подлежит амплификации. Установив это, синтезируются два коротких ДНК-зонда (праймеры) с таким расчетом, чтобы они могли вступить в гибридизацию с комплементарными участками ДНК-мишени (рис. 4). Тест-система включает ДНК, выделенную из исследуемого материала, праймеры, дезоксирибонуклеотиды (в виде трифосфатов) и термостабильную ДНК-полимеразу (продукт термофильных бактерий Thermus aquaticus). Реакционная смесь подвергается повторным циклам нагревания-охлаждения, во время которых совершается денатурация ДНК (нагревание), гибридизация/отжиг праймеров (охлаждение) и синтез ДНК. В первом цикле происходит дупликация фрагмента ДНК, ограниченного праймерами, причем каждая из разобщенных ДНК-нитей служит матрицей для синтеза второй, комплементарной нити ДНК, начиная от связанного праймера. При синтезе (полимеризации) очередной ДНК-нити генерируется и новый участок для связывания праймера. Это означает, что любая вновь синтезированная нить становится матрицей для последующих синтетических циклов, инициируемых праймерами. Число копий амплифицируемого ДНК-фрагмента (а, следовательно, и ДНК-матриц для последующего синтеза) с каждым циклом удваивается, нарастая в геометрической прогрессии (отсюда — цепная реакция). В этом отношении ПЦР аналогична естественной репликации ДНК в делящихся клетках, но в ПЦР экспоненциальный синтез ДНК лимитирован фрагментом, на котором фиксирован искусственно синтезированный праймер. Продукты ПЦР (ампликоны) идентифицируют при помощи гелевого электрофореза или путем гибридизации с ДНК-пробой, комплементарной амплифицируемому фрагменту ДНК.

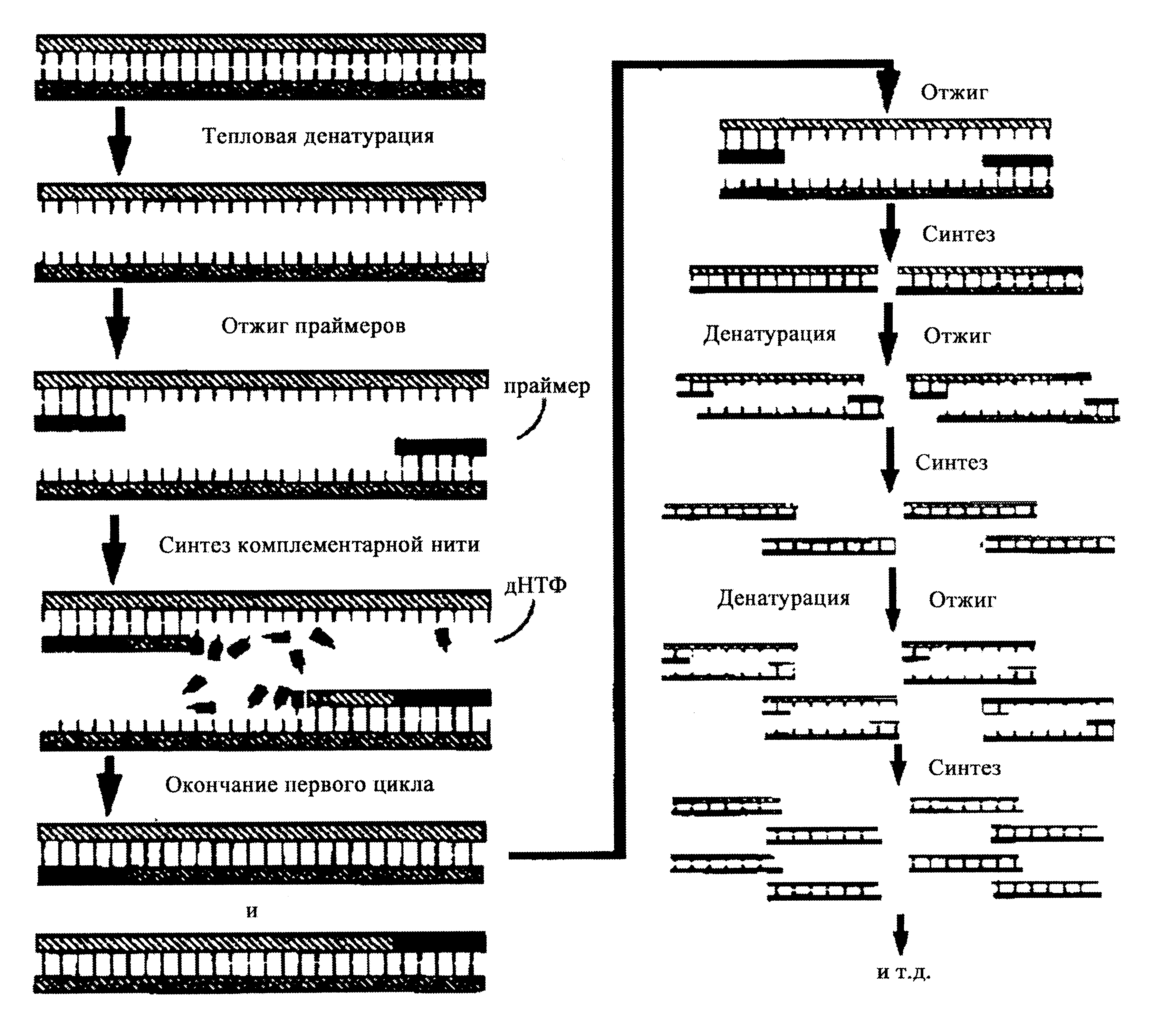
Достоинством ПЦР является ее феноменальная чувствительность: теоретически она может быть возбуждена единственной копией ДНК-мишени. Но это и главный недостаток ПЦР как клинико-диагностического теста: он создает опасность ложноположительных результатов за счет минимальных загрязнений посторонними ДНК, особенно в лабораториях, регулярно выполняющих однотипные анализы. Впрочем, это чисто техническое возражение, которое при соблюдении надлежащего режима не может повлиять на диагностические возможности ПЦР. Более серьезные опасения возникли из-за ложнонегативных результатов, связанных с присутствием в исследуемом материале ингибиторов полимеразы. К счастью, с этим сталкиваются нечасто, но для надежности требуются специальные контроли.
Принцип специфической амплификации нуклеиновых кислот, заложенный в классическом варианте ПЦР, определил ряд новых направлений в диагностике инфекционных заболеваний. Отметим, например, выявление штаммов, резистентных к антибиотикам. Это достигается определением кодирующих генов, которые метят при помощи праймеров к участкам ДНК, отвечающим за данный признак. Для накопления вирусных РНК-генов ПЦР применяется после обратной транскрипции РНК в ДНК, что достигается при помощи ретровирусной транскриптазы. Широко используется лигазная цепная реакция (сшивание меченого и немеченого фрагментов гена под действием ДНК-лигазы), прямая репликация рибосомальных РНК (при помощи РНК-праймеров и РНК-полимеразы) и др. Словом, золотой эталон, которым считается выделение и идентификация чистой культуры возбудителей, выглядит уже не столь абсолютным на фоне некультуральных приемов, основанных прежде всего на выявлении генетических маркеров микроорганизмов.
Выявление иммунных сдвигов в организме хозяина
Присутствие микроорганизма создает конфликтную ситуацию, символом которой является иммунный ответ — образование антител и формирование клеточного иммунитета.
Антитела. Одним из универсальных признаков инфекции служат результаты серологического (иммунохимического) анализа, отражающего гуморальную реакцию хозяина на воздействие антигенов возбудителя. Серологический анализ означает исследование сыворотки крови (лат. serum — cыворотка), которая служит субстратом для накопления антител. Способы определения антител прошли ту же эволюцию, о которой говорилось выше в связи с иммунохимическими реакциями по выявлению антигенов. Первые методы, основанные на констатации внешних эффектов (агглютинация, преципитация, нейтрализация, связывание комплемента), почти целиком уступили место реакциям, где о положительном результате судят по связыванию меченых пероксидазой или щелочной фосфатазой антител против иммуноглобулинов человека (рис. 5).

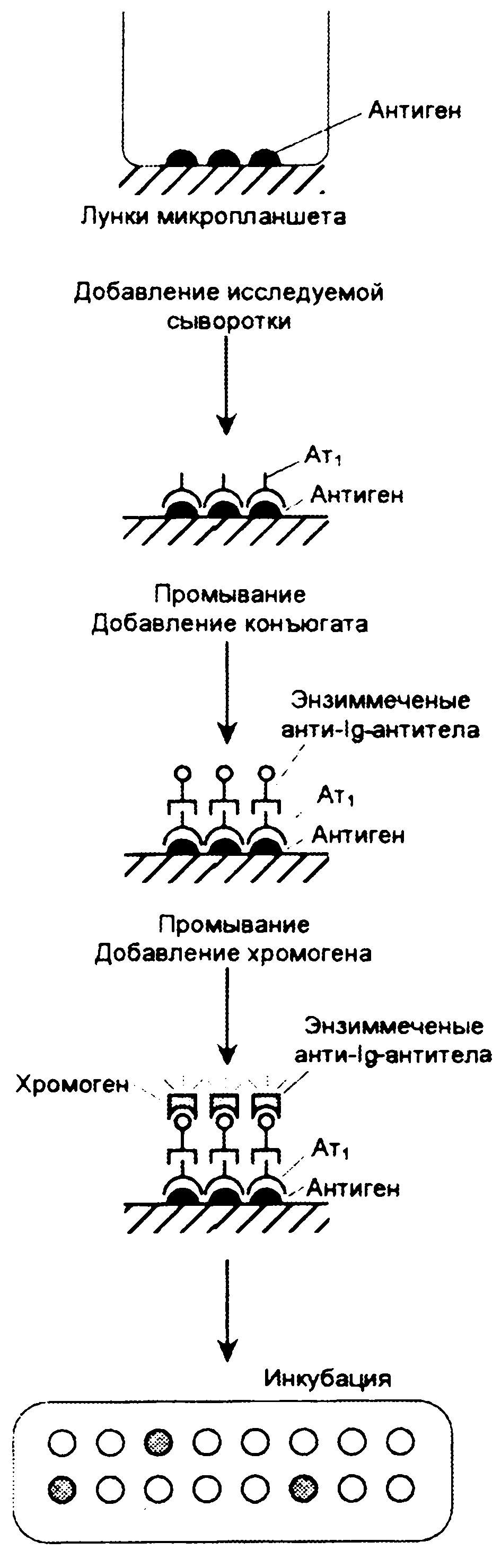
К недостаткам серологического анализа относится ретроспективность, т.е. невозможность ранней диагностики острых инфекций. Это связано с тем, что для образования антител требуется время, которое составляет от двух недель до нескольких месяцев. Поэтому серологический метод позволяет судить о том, что данная инфекция произошла не сейчас, а в прошлом, и именно там надо искать источник и причину заражения.
Еще одной задачей, которую приходится решать, служит расшифровка того, являются ли антитела результатом текущего заболевания или они возникли раньше, от инфекции, случившейся месяцы или годы тому назад. Для ответа на этот вопрос используется то, что можно назвать количественной и качественной сероконверсией. Количественная конверсия основана на колебаниях титров специфических антител в динамике заболевания. Обнаружение значительного подъема антител говорит об участии патогена в происхождении болезни. О качественной сероконверсии судят по изотипу (классу) выявляемых антител, а также по спектру антител к различным антигенам возбудителя. В первом случае обычно измеряют IgM-антитела против подозреваемого микроба, которые при большинстве инфекций появляются первыми, исчезая через несколько недель или месяцев после начала болезни.
Спектр антител выявляют в реакции иммуноблоттинга (от англ. blot — промокать, пятно). Она сочетает в себе иммуноферментный анализ с электрофорезом и предназначена для изучения профиля гуморального ответа к антигенным смесям. Это имеет важное значение для исключения ложноположительных результатов и применяется в диагностике таких инфекций, как гепатит С и СПИД.
Принцип метода показан на рис. 6. Смесь антигенов, экстрагированных из исследуемого объекта (в данном случае вируса иммунодефицита человека — ВИЧ), разделяют на фракции при помощи электрофореза в полиакриламидном геле. Гель покрывают (промокают) нитроцеллюлозной мембраной, воспроизводя на ней точную копию белковых фракций, полученных в полиакриламидном блоке. На заключительном этапе полоски (стрипы) нитроцеллюлозы анализируют в реакции ИФА с исследуемой сывороткой. Количество выявленных фракций (пятен) отражает спектр антител к антигенам анализируемой смеси. Наличие антител к двум и более антигенам предполагаемого возбудителя не может быть случайным, подтверждая диагноз заболевания.

![Рис. 6. Иммуноблоттинг. Определение спектра анти-ВИЧ-антител. Этап 1: Вирусные антигены [белки (p), гликопротеины (gp)] подвергаются электрофорезу в блоке полиакриламидного геля. Это ведет к разделению антигенов на фракции по молекулярной массе (мол. масса в кДа отмечена цифрами). Этап 2: фракции антигена переносят при помощи электрофореза на нитроцеллюлозную мембрану, которую разрезают на полоски (стрипы). Этап 3: стрипы инкубируют с исследуемой сывороткой и затем отмывают от несвязавшегося материала. Этап 4: стрипы инкубируют с энзиммечеными антителами против иммуноглобулинов человека, отмывают. Этап 5: добавляют хромогенный субстрат и отмечают число окрашенных фракций (пятен). Они совпадают с зоной локализации антигенов, которые прореагировали с антителами исследуемой сыворотки. В данном случае сыворотка содержит антитела против gp41, p24 и р31. Это доказывает диагноз ВИЧ-инфекции](https://medread.ru/wp-content/uploads/6_006_.png)
Наибольшее признание получили кожные тесты, выявляющие гиперчувствительность (аллергию) IV типа к микробным продуктам. Таких инфекций немного, так как внутрикожное введение микробных ингредиентов чревато большим числом ложноположительных и ложноотрицательных реакций. Отметим лишь один пример — пробы с туберкулином, белковыми дериватами туберкулезной палочки. Правильная оценка туберкулиновых реакций (ее проводят через 48—72 ч) оказывает существенную помощь в оценке туберкулезного процесса у индивидуальных больных, позволяя своевременно принимать профилактические и лечебные меры.
* * *
Современная микробиология оснащена аналитическими приемами, которые позволяют решать комплекс проблем, связанных с диагностикой, лечением и профилактикой инфекционных заболеваний. Поражает техника иммунохимических реакций, в которых используются меченые антитела. По своей точности и чувствительности они близко стоят к культуральному методу, но превосходят его по универсализму. Еще больше перспектив у реакций, основанных на молекулярном выявлении генных продуктов микроорганизмов. Уже сегодня результаты многих из них соответствуют классическому тестированию с использованием трудоемких методов культурального анализа. В этом будущее микробиологического анализа, целью которого является высококачественная экспресс-диагностика инфекций, т.е. быстрая индикация возбудителей без выделения чистой культуры.
Стафилококки
Не так страшно стадо зверей, как один лиходей.
- Золотистый стафилококк :
- местные и системные инвазии;
- факторы инвазии;
- резистентность к антибиотикам;
- специфические интоксикации;
- иммунитет.
- Болезнетворность «непатогенных» стафилококков.
- Внутрибольничные инфекции.
Стафилококки принадлежат к группе наиболее популярных объектов медицинской микробиологии. Будучи симбионтами человека и животных, они не всегда ограничивают свои взаимоотношения с хозяином рамками безвредного сожительства. Местом нормального обитания стафилококков служат кожа, ее придатки и слизистые оболочки (чаще носа). Именно здесь обычно возникают первичные очаги инфекции, которые развиваются по типу пиогенных инвазий. Но этим далеко не исчерпывается спектр стафилококковых заболеваний. Он гораздо шире, так как в любой части тела, куда попадает стафилококк, может сформироваться патологический процесс. Всю жизнь человек проводит в среде, обсемененной стафилококками, и лишь редкие люди избегают стафилококковых поражений, хотя, к счастью, тяжелые инфекции встречаются редко.
Стафилококки — грамположительные шаровидные бактерии, 0,5—1,5 мкм в диаметре, неподвижны, образуют скопления, напоминающие гроздья винограда (греч. staphylos — гроздь; рис. 1).

Этих двух признаков (окраска по Граму и морфология) достаточно, чтобы отличить стафилококки от других бактерий. Для дифференциации видов требуется знать биохимические особенности, структурные детали клеточной стенки, ферментативную активность и пр. За небольшим исключением (группа облигатно анаэробных стафилококков-пептококков) все стафилококки — факультативные анаэробы, способные не только выживать в присутствии кислорода, но и утилизировать его в реакциях, обеспечивающих энергию. Стафилококки продуцируют каталазу (фермент, разрушающий перекись водорода), и это в известной мере защищает их от высокотоксичных производных кислорода. Кроме того, в аэробных условиях стафилококки синтезируют каротиноидные пигменты (этим, в частности, объясняется цвет колоний), которые также нацелены против оксидантов. Устойчивость к биологически активным формам кислорода служит одним из факторов резистентности стафилококка в условиях пиогенных инвазий. Стафилококки не образуют спор, но из бесспоровых бактерий они относятся к числу наиболее устойчивых во внешней среде. Отметим и способность стафилококков расти на простых питательных средах. Это значительно упрощает микробиологическую диагностику и другие процедуры, связанные с культивированием бактерий. Не случайно стафилококк был в числе первых бактерий, изолированных от человека. Это сделал Бекер в 1883 г. после того, как Клебсом, Кохом и Пастером была высказана и экспериментально обоснована идея об инфекционной природе нагноений. Название «стафилококк» предложено Огстоном в 1881 г.
Устойчивость и неприхотливость стафилококков проявляется и в том, что они размножаются в присутствии высоких концентраций хлорида натрия (до 10—15%). Этот признак используют при изготовлении дифференциально-диагностических сред: в них добавляют избыток поваренной соли для подавления роста других бактерий. Галофильность золотистого стафилококка имеет и эпидемиологическое значение, способствуя обсеменению энтеротоксигенными штаммами пищевых концентратов (см. ниже). Отмечая устойчивость стафилококков, обратим внимание на их парадоксальную чувствительность к анилиновым красителям, например к кристаллвиолету (1:500000) и к брильянтовому зеленому (1:1000000). Это давно используется в лечении стафилококковых пиодермий.
Известно около 30 видов стафилококка, которые объединены в родовой таксон (Staphylococcus) семейства Micrococcaceae. Из них 14 являются симбионтами человека. Различные виды связаны умеренным генетическим родством: в жестком (рестриктивном) режиме гомология их ДНК не превышает 20%. Человек и животные могут инфицироваться одними и теми же видами, но некоторые стафилококки выделяются преимущественно от животных или людей. У человека доминируют три вида — S. aureus, S. epidermidis и S. saprophyticus. Почти все медицинские проблемы связаны с золотистым стафилококком (S. aureus), наделенным многими атрибутами патогенности. Именно о нем принято говорить как о патогенном стафилококке, отдавая должное его значимости в патологии. Впрочем, учитывая широкое носительство S. aureus здоровыми людьми и зависимость стафилококковых инфекций от дополнительных обстоятельств, золотистый стафилококк следует скорее отнести к разряду условно-патогенных бактерий. Иными словами, стафилококковые заболевания в большинстве вторичны, т.е. протекают по типу пиогенных осложнений.
Значение других стафилококков за редким исключением ограничено проблемой госпитальных (внутрибольничных) инфекций. Реализация их весьма условной патогенности связана с инвазивностью современных диагностических и лечебных процедур, которые способствуют внедрению бактерий, широко представленных в микрофлоре человека и окружающей среде.
Если не считать общности тинкториальных свойств, морфологии и ряда базисных физиологических признаков, то сходство между золотистым и «непатогенными» стафилококками невелико. Это относится и к факторам вирулентности, многие из которых есть только у S.aureus и используются в таксономике. Но исторически сложилось так, что видовой эпитет самого опасного стафилококка отражает патогенетически не ясный и вариабельный признак — пигментообразование. Колонии золотистого стафилококка окрашены в желтый или оранжевый цвет, что и послужило поводом для термина «золотистый» (aureus). Альтернативой некогда был «белый» стафилококк — таксон, который позже распался на дискретные виды, включая S.epidermidis и S.saprophyticus.
Золотистый стафилококк (S. aureus)
Внутривидовая классификация
Отличить S. aureus от других стафилококков нетрудно, тем более что его культуральные особенноcти легко усилить при помощи дифференциально-диагностических сред, выявляющих активность ферментов (липаза, дезоксирибонуклеаза, стафилокиназа) или токсинов (гемолизины) (рис. 2). Изучение культуральных признаков, мазок из колонии, окрашенный по Граму, и тест на свертывание плазмы (из стафилококков человека только S. aureus обладает коагулазной активностью) фактически исчерпывают рутинный микробиологический анализ. Впрочем, иногда приходится решать еще две задачи: определение чувствительности к антибиотикам и внутривидовое типирование для выяснения закономерностей распространения инфекции (источник заражения, пути передачи).
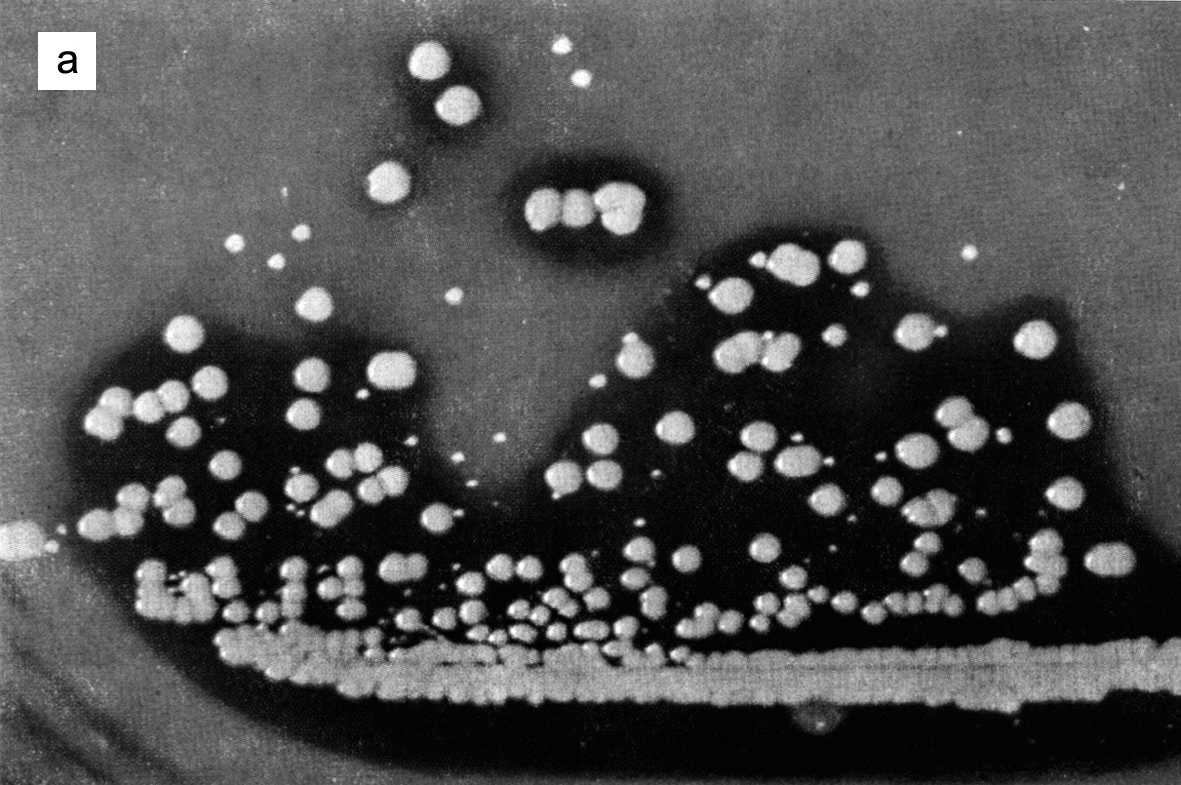
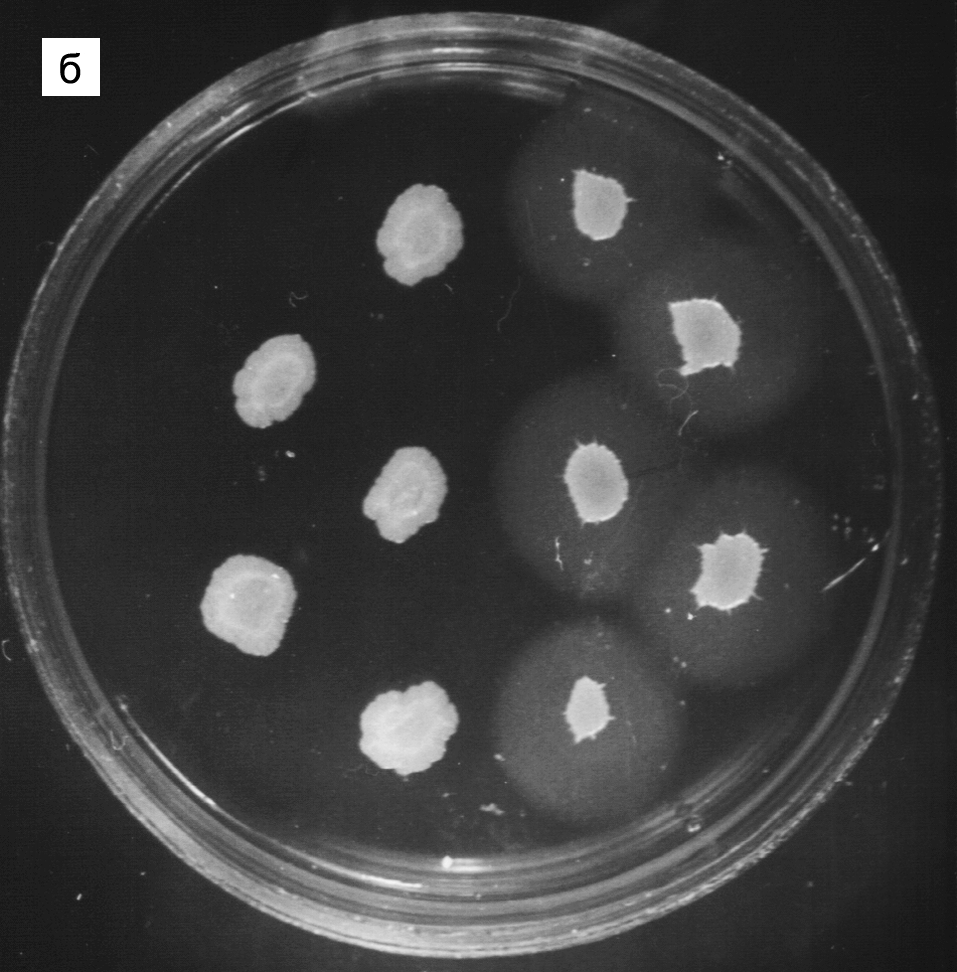


Рис. 2. Колонии стафилококков на дифференциально-диагностических средах. На кровяном агаре (а) колонии S.aureus окружены зоной гемолиза; колонии S.epidermidis — мелкие, негемолитические. На желточно-солевом агаре (б) колонии S. aureus окружены мутным орелом расщепленных липидов (результат высокой липазной активности)
Серологическая дифференцировка штаммов не является общепризнанной. Стафилококки обладают сложным и непостоянным набором поверхностных антигенов и использовать их для типирования бактерий не удалось. По капсульному полисахариду золотистый стафилококк может быть разделен на 11 серотипов, однако этого явно недостаточно для эпидемиологической практики. Штаммы S. aureus можно различить по генотипу или по спектру секретируемых белков, но чаще применяется фаготипирование c помощью международного набора бактериофагов (табл. 1).
Т а б л и ц а 1
Фаготипирование золотистого стафилококка
| Фагогруппа | Фаги (фаготип, или фаговар) |
| I | 29, 52, 52A, 79, 80 |
| II | 3A, 3C, 55, 71 |
| III | 6, 42E, 47, 53, 54, 75, 77 |
| IV | 42D |
| Вне групп | 81, 94, 95, 96 |
Одна и та же культура может лизироваться разными фагами, из чего складывается профиль фагочувствительности (фагомозаика) штаммов. Так как число подобных спектров велико и на практике их трудно учитывать, фаги объединены в группы. Каждый штамм относится к одной из фагогрупп и соответствующему фаготипу; предпочтение отдают фагам с наибольшей литической активностью. Совпадение фагомозаики у штаммов, изолированных от разных больных, говорит об общем источнике инфекции.
Экология
Золотистый стафилококк принадлежит к числу широкораспространенных бактерий. Он часто выделяется от здоровых людей и с этой точки зрения может рассматриваться как факультативный компонент нормальной микрофлоры. Контакт с ним неизбежен, и уже в первые дни жизни (в родильном доме) практически все новорожденные подвергаются контаминации. Повышенный уровень инфицирования наблюдается и в обычных медицинских стационарах, причем как среди больных, так и у обслуживающего персонала. Чаще всего золотистый стафилококк локализуется на слизистой оболочке носа и носоглотки. Примерно 20% людей не поддерживают носительства. Для большинства людей (около 60%) носительство транзиторно: они освобождаются от стафилококка через несколько дней или недель и, если заражаются вновь, то уже другим штаммом. Но встречаются и упорные носители (их около 20%), у которых тот же штамм выделяется месяцы и даже годы. Это наиболее вероятный источник инфицирования окружающих, особенно при высокой обсемененности носителя (эпидемически опасным считается присутствие более 10 млн. бактерий в 1 мл назального секрета).
Борьбе с носительством золотистого стафилококка отдано немало сил. Этот вопрос особенно важен для медицинских учреждений, где угрозе заражения подвергаются ослабленные больные и дети. Однако реальных успехов и общепризнанных рекомендаций добиться не удалось. Борьба со здоровым носительством — это по сути борьба с нормальной микрофлорой, что не всегда проходит бесследно для природных микробиоценозов, провоцируя развитие дисбиотических состояний. Об этом следует помнить и санировать лишь злостных носителей, т.е. лиц, упорно выделяющих большие дозы бактерий. Появились надежды, связанные с местными аппликациями антибиотика мупироцина (природного продукта Pseudomonas fluorescens), который неплохо справляется с интраназальным носительством золотистого стафилококка. Он быстро метаболизируется сывороточными ферментами, а потому должен применяться местно. Это первый препарат, действующий на тРНК-синтетазы (в том числе на метациллинрезистентные клоны золотистого стафилококка), и есть надежда, что будут получены аналоги мупироцина для внутреннего использования.
Вторая по значимости экологическая ниша для S. aureus — кожа, особенно участки с повышенной влажностью (у 10—20% людей золотистый стафилококк выделяется из промежности). Колонизация кожи создает основу для ее пиогенных инвазий — самой распространенной стафилококковой инфекции. В небольших количествах золотистый стафилококк присутствует в толстом кишечнике и влагалище. Изредка это служит причиной местных поражений или системных интоксикаций.
В основе клинического разнообразия стафилококковой патологии лежит способность S. aureus 1) вызывать пиогенную инвазию кожи и ее придатков, 2) проникать в кровь и индуцировать пиогенные (гнойно-деструктивные) поражения внутренних органов, 3) вызывать неспецифическую (септическую) и 4) специфические интоксикации.
Местные инвазии
Чаще всего S. aureus поражает кожу с развитием пиодермий — не только самой распространенной формы стафилококковой инфекции, но и пиогенных инвазий вообще. Слизистые оболочки поражаются редко, и в этом отношении стафилококк отличается от стрептококка (Str. pyogenes), который чаще инфицирует слизистые оболочки. Отсюда сравнительно небольшое значение золотистого стафилококка при ангинах, отитах, гайморитах и первичных поражениях бронхолегочного аппарата. Стафилококковая инфекция выступает здесь как фактор, осложняющий течение основного патологического процесса.
Сущность любой пиогенной инвазии — в столкновении между бактериями, запускающими повреждение ткани, и лейкоцитами (нейтрофилами), мобилизуемыми из крови благодаря воспалительной реакции. Если возбудитель не может быть быстро уничтожен, формируется патологический (т.е. клинически значимый) процесс — очаг гнойного воспаления. Это наблюдается при большой инфицирующей дозировке, повышенной вирулентности возбудителя, дефектах в системе опсонофагоцитарного иммунитета. Установлено, например, что искусственная задержка выхода нейтрофилов в зону введения стафилококка резко снижает концентрацию бактерий, необходимую для активизации пиогенного процесса: происходит опережающее размножение бактерий, которые успевают накопить силы для сопротивления «нерасторопным» фагоцитам.
Типовой манифестацией стафилококковой инвазии (независимо от локализации) является нарыв, или абсцесс, т.е. отграниченный очаг гнойного воспаления (лат. abscedo — отделяю, отсекаю). При пиодермиях изначально поражаются придатки кожи (волосяные фолликулы, сальные и потовые железы).
По месту и характеру поражения различают следующие варианты:
1) фолликулит — гнойное воспаление (пустула) волосяного фолликула;
2) сикоз — гнойное воспаление волосяных фолликулов лица (усы, борода), реже — подмышечных впадин, лобка;
3) фурункул — гнойное воспаление волосяного фолликула, захватывающее окружающие ткани;
4) ячмень — гнойное воспаление сальной железы (железы Цейса) волосяного фолликула ресницы;
5) карбункул — слияние нескольких фурункулов;
6) гидраденит — гнойное воспаление апокриновых потовых желез;
7) импетиго — поверхностное поражение кожи типа гнойничковой сыпи, обычно в ассоциации с пиогенным стрептококком;
8) синдром ошпаренной кожи (син. — эксфолиативный дерматит, генерализованная пузырчатка, болезнь Риттера) — отслоение эпидермиса на значительной поверхности туловища и конечностей после слияния и изъязвления пузырьков с серозным содержимым. Это редкое заболевание, связанное с действием эксфолиативного токсина (см. ниже), обычно поражает детей в возрасте до 12 мес, чаще — в первые дни после рождения. Местным проявлением синдрома является пузырчатка (пемфигус); обычная локализация — паховые складки, подмышечные впадины, кожа живота, шеи.
Проникновению бактерий в железистые протоки способствуют микротравмы кожи, например от трения одежды. Поэтому фурункулы появляются на шее, затылке, пояснице, в подмышечной области. Вероятность инвазии возрастает при несоблюдении правил гигиены, что ослабляет самоочищение кожи от бактерий. Нередко стафилококк внедряется с инородными телами, например с занозами. В таких случаях для возбуждения патологического процесса требуется гораздо меньше бактерий. Так, если для получения микроабсцесса достаточно ввести с шовным материалом всего 100 клеток золотистого стафилококка, то при инъекции взвеси того же штамма требуется около 1 млн. бактерий. Возможно, в таких случаях проявляется способность S. aureus к образованию биопленки на инородных материалах. Это связано с выделением адгезивного полисахарида, поли-n-сукцинил-b-1,6 глюкоамина, который способствует прилипанию стафилококков друг к другу и к другим (чужеродным) субстратам.
Впрочем, в отличие от других бактерий S. aureus способен поражать и неповрежденную кожу, проникая через устья волосяных фолликулов или протоки потовых желез. При поражении апокринных потовых желез (в отличие от обычных, эккринных желез, они открываются не на поверхность кожи, а в волосяные фолликулы) возникает разновидность фурункулеза — гидраденит. Так как апокринные железы локализуются в определенных участках тела и развиваются к моменту половой зрелости, гидраденит встречается только у взрослых людей, чаще в подмышечной области.
С патогенетической точки зрения к стафилококковым пиодермиям примыкает мастит (или грудница) кормящих матерей. В этом случае абсцесс формируется в молочной железе при внедрении возбудителя в железистые протоки через микротравмы при грудном кормлении. Источником заражения обычно являются новорожденные, ротовая полость которых с первых дней жизни обильно контаминирована золотистым стафилококком. Кстати, сходное заболевание наблюдается у коров; здесь бактерии заносятся руками доильщиц и причиной часто служат стрептококки (Str. agalactiae).
Системные инвазии
Стафилококковые абсцессы могут осложняться бактериемией (заражением крови), которая угрожает пиогенными метастазами, порождающими новые очаги гнойного воспаления. Возникает разновидность сепсиса — септикопиемия, патогенез которой складывается из истощающей интоксикации и нарушения органных функций. Изредка доминирует интоксикация, и процесс, обретая скоротечную (фульминантную) форму, завершается смертельным исходом через несколько дней.
Причиной септикопиемии служит фрагментация инфицированных тромбов протеолитическими ферментами гноя, создающая опасность эмболии. Внутри микротромбов бактерии неуязвимы для бактерицидных факторов крови и вместе с ними оседают в сосудистом русле, получая возможность для продолжения септического процесса. Генерализация инфекции наиболее вероятна у ослабленных больных с обширной зоной местной инвазии (например, при ожогах). Но иногда она возникает на фоне незначительных местных поражений, от которых, казалось бы, трудно ожидать cерьезных последствий.
Впрочем, при одной из локализаций пиогенного процесса вероятность метастатических осложнений резко возрастает. Речь идет о нарывах лица в так называемом опасном треугольнике — области с основанием вдоль верхней губы и вершиной над переносицей (рис. 3). К особенностям этой зоны относятся слабое развитие соединительной ткани, отсутствие клапанов в венах, напрямую связанных с мозговыми синусами, постоянное движение мимической мускулатуры. Это ослабляет механические барьеры, создавая угрозу смертельных инфекционных поражений головного мозга (энцефалит) и его мягкой оболочки (менингит). Поэтому область опасного треугольника нельзя травмировать, тем более выдавливать нарывы, провоцируя проталкивание гнойного содержимого в сосудистое русло.


Опасность метастатических абсцессов угрожает любому органу, но чаще всего инфицируется костный мозг. Его заражение, захватывающее костную ткань (остеомиелит), нередко служит единственным или первым признаком генерализации пиогенного процесса. Источником инфекции чаще являются очаги пиодермии, откуда стафилококк распространяется гематогенным путем — гематогенный (метастатический) остеомиелит. Эта почти уникальная способность золотистого стафилококка давно подмечена микробиологами. Еще Пастер метко назвал остеомиелит фурункулом костного мозга. Без антибактериальной терапии события развиваются по следующей схеме. Нагноение костного мозга переходит в субпериостальный абсцесс, который распространяется вдоль диафиза либо прорывается через надкостницу, поражая прилегающую мышечную ткань. Тромбоз сосудов ведет к некрозу костной ткани и секвестрации мертвых фрагментов, выходящих с гноем через свищевые ходы. Будучи следствием внутрисосудистой бактериальной инвазии (бактериемии), очаг остеомиелита сам становится угрозой септикопиемических осложнений. Процесс сопровождается образованием биопленки и нередко обретает затяжное течение. Тогда угроза септических осложнений становится постоянной.
Массивная бактериемия создает опасность пиогенного поражения клапанов сердца — самого страшного осложнения стафилококковой бактериемии. Чаще колонизируются органически измененные клапаны (после хирургического вмешательства, при ревматизме), причем достаточно небольшого повреждения, чтобы процесс стал необратимым. Изредка поражаются и нормальные клапаны (особенно аортальные). В клеточной стенке S. aureus обнаружены компоненты, которые обеспечивают адгезию бактерий на эндотелиоцитах, а, следовательно, и на поверхности эндокарда.
Много написано о стафилококковой пневмонии. Она протекает по типу пиогенной деструкции легких, завершаясь быстрой смертью или абсцедированием. Абсцессы прорываются в плевральную полость, приводя к накоплению в ней гнойного экссудата (эмпиема). Поражение развивается как результат внутрипульмонального заноса бактерий из крови либо как экзогенная инфекция на фоне предрасполагающих факторов (грипп, корь, коклюш, муковисцидоз, оперативные вмешательства, иммунодефицитные состояния). Экзогенные стафилококковые пневмонии наиболее опасны для детей с острой респираторной патологией.
Стафилококковая инвазия чревата тяжелой интоксикацией, вплоть до шока. Это финал септического синдрома, патогенез которого определяется суммарным эффектом микробных и вторичных токсинов, выделяющихся при разбалансированной гиперстимуляции эффекторов местного и системного воспаления. Компоненты клеточной стенки грамположительных бактерий (тейхоевые кислоты, пептидогликан) обладают эндотоксиноподобным действием, активируя комплемент, кининовую и свертывающую систему, продукцию цитокинов, эйкозаноидов, эндорфинов, окиси азота и пр. Это ведет к расширению патогенетического каскада стафилококковой инвазии и стандартным проявлениям фатального сепсиса (см. «Сепсис»).
Факторы инвазии
Эволюция пиогенных инвазий определяется конфронтацией двух процессов — жизнедеятельности бактерий и воспалительной реакции организма. Давний спор о том, что здесь главное — хозяин или паразит, не может быть решен однозначно. С открытием цитокинов и других медиаторов-эффекторов воспаления стало очевидным, что воспалительные реакции сопровождаются выделением множества факторов с многоплановой (местной и системной) биологической активностью, которые не только санируют, но и вызывают тяжелые осложнения, формально связываемые с бактериями. Это особенно характерно для септических (гнойных) инфекций, возбудители которых нередко лишены прямых деструктивных потенций. На этом основании в патогенезе пиогенных инвазий следует дифференцировать роль микробных факторов и эндогенных эффекторов воспаления. Эта позиция блестяще отражена в парадоксе Л. Пастера, справедливом для всех оппортунистических инфекций: «Микроб — ничто, субстрат (т.е. организм — А.М.) — все». Помня об этом, мы, тем не менее, сконцентрируем внимание на микробзависимых факторах стафилококковых инвазий.
Адгезия. Наиболее общим понятием, характеризующим устойчивость к инфекции, является колонизационная резистентность — способность противостоять закреплению (адгезии) и размножению микробов в тканях.
Можно выделить два уровня колонизационной резистентности, которые необходимо преодолеть инвазивным бактериям. Один из них — первичная, или базисная резистентность. Ею в той или иной степени обладают все ткани, она предсуществует до внедрения возбудителя, носит пассивный характер и определяется суммой резидентных (гуморальных и клеточных) факторов, которые препятствуют адгезии микробов, обладают биостатической и биоцидной активностью. На втором уровне колонизационная резистентность носит реактивный характер. Она развивается в ответ на микробную инвазию, нацелена на ее ограничение, уничтожение инфекта и восстановление местного гомеостаза. Эффекторы концентрируются в зоне поражения благодаря воспалительной реакции, которая выступает в роли аварийного рычага антиинвазивной защиты. Именно в очаге воспаления достигается функциональная стыковка гуморальных и клеточных механизмов антимикробного иммунитета, неспецифических и специфических факторов защиты. При пиогенных инвазиях центральное место принадлежит нейтрофилам, хотя их реакции — лишь вершина событий, инициирующих и развивающих воспалительный процесс.
На основе этих общих замечаний проанализируем свойства золотистого стафилококка, которые имеют отношение к патогенезу пиогенных инвазий. Пожалуй, ни один из видов бактерий не может конкурировать с ним по числу факторов с потенциальной болезнетворностью. Это отражает сложность патогенеза стафилококковых инфекций и обусловливает стремление отыскать истинную причину их многообразия.
Желание понять главное вполне естественно, но формальное перечисление микробных факторов вирулентности (особенно в приложении к условно-патогенным бактериям) оправдано лишь в дидактических целях. Следует помнить, что патогенность/вирулентность — поливалентный признак, реально раскрывающийся лишь в диалектике инфекционного процесса.
Необходимо отметить, что S. aureus обладает сложным набором поверхностных белков-адгезинов, которые обеспечивают его прикрепление к различным биологическим субстратам. Они связываются с муцином слизистых оболочек, протеогликанами соединительной ткани и эндотелиоцитов, фиксируются на белках экстрацеллюлярного матрикса (коллагене, фибронектине, ламинине, костном сиалопротеине), рецептируют растворимые фибронектин, витронектин, фибриноген и другие сывороточные белки. В сочетании с транскрипционной регуляцией адгезинов и связанной с ней адаптивной (фазовой) изменчивостью адгезивных свойств это объясняет почти неограниченные возможности золотистого стафилококка в колонизации тканей хозяина.
Липаза (лецитовителлаза). Как уже говорилось, S. aureus чаще всего вызывает абсцедирование кожи, проникая в ее придатки. Следует отметить два свойства, которые определяют способность золотистого стафилококка внедряться в волосяные фолликулы, сальные и потовые железы и персистировать в них. Во-первых, он обладает мощной липазной активностью, позволяющей разрушать сальную пробку в устье волосяного мешочка, получая одновременно продукты питания. Во-вторых, он устойчив к высоким концентрациям хлорида натрия и жирным кислотам, что обеспечивает выживание в секретах потовых и сальных желез. Жирные кислоты, которые образуются при бактериальном расщеплении нейтральных жиров, холестерина и других липидов, усиливают воспалительную реакцию на микробную инвазию. Получая развитие, она переходит в патологический процесс — очаг гнойного воспаления.
Капсула. Стабилизация, а тем более эволюция инфекции зависит от микробных факторов, оказывающих сопротивление фагоцитозу. Они срабатывают на уровне прямых контактов с фагоцитами, блокируют опсонины, повреждают фагоцитирующие клетки, способствуют выживанию внутри фагоцитов. Антифагоцитарные факторы золотистого стафилококка приведены в табл. 2. Некоторые из них дополнительно прокомментированы ниже.
Способностью к образованию капсулы обладают большинство штаммов золотистого стафилококка, хотя в классическом (заметном при микроскопии) виде она встречается редко. Описано 11 серотипов капсульного полисахарида. Из них лишь серотипы 1 и 2 имеют обильную капсулу, которая проявляется при исследовании колоний (слизистая консистенция). Заметим, что они редко выделяются при клинической патологии.
Т а б л и ц а 2. Факторы инвазии S. aureus
| Фактор | Эффект |
| Капсула (немногие штаммы) | Подавление взаимодействия с фагоцитами |
| Капсулоподобный экзополисахарид (более 90% штаммов) | То же |
| Коагулаза | Образование сгустков фибрина, препятствующих фагоцитозу (псевдокапсула) |
| Хлопьеобразующий фактор | То же |
| Стафилолизины (гемолизины): альфа-токсин бета-токсин гамма-токсин дельта-токсин |
Лизис нейтрофилов и макрофагов То же -«- -«- |
| Лейкоцидин (2—3% штаммов) | Лизис нейтрофилов |
| Каталаза | Подавление миелопероксидазной активности (разрушение перекиси водорода) |
| Каротиноидные пигменты | Инактивация бактерицидных форм кислорода |
| Антифагин (природа неизвестна) | Подавление взаимодействия с нейтрофилами (механизм не известен) |
| Белок А | Подавление опсонической активности антител (экранирование Fc-фрагмента IgG) |
| Липаза (лецитовителлаза) | Разрушение жировой пробки в устье волосяных фолликулов |
| Пептидогликан | Устойчивость к лизоциму |
| Гиалуронидаза | Расширение зоны инфекции за счет разрушения соединительной ткани |
| Стафилокиназа | Разрушение фибриновых сгустков с образованием инфицированных микротромбов |
| Коллагеназа | Повышение проницаемости тканей за счет разрушения коллагена |
| Дезоксирибонуклеаза | Расщепление ДНК, разжижение гноя |
| Устойчивость к высоким концентрациям хлорида натрия и жирным кислотам | Размножение в потовых и сальных железах |
Инкапсулированные штаммы резистентны к фагоцитозу и легко убивают мышей при внутрибрюшинном введении; предварительная иммунизация убитой вакциной из капсульного штамма защищает от смертельной инфекции. Однако, учитывая ограниченное распространение, капсулу нельзя считать универсальным фактором патогенности S. aureus, хотя генетически закрепленная готовность к капсулообразованию позволяет думать о возможности проявления этого признака в условиях естественной инфекции.
Коагулаза. Все штаммы золотистого стафилококка обладают почти уникальной способностью индуцировать образование фибрина, свертывая плазму. Плазмокоагулирующая активность — главный видовой признак S. aureus, который отлично коррелирует с вирулентностью и продукцией других факторов патогенности. Коагулаза действует на протромбин с образованием тромбиноподобного комплекса, который переводит фибриноген в фибрин. Реакция не зависит от Са2+, а потому происходит в хелатированной (например, цитратной) плазме.
Кроме секретируемой коагулазы, золотистый стафилококк обладает еще одним фибринпродуцирующим эффектом: он зависит от хлопьеобразующего фактора. Такого рода «связанная» коагулаза кроме механизма действия отличается от «свободной» по антигенной специфичности. Фиксируя из плазмы мономеры фибрина, хлопьеобразующий фактор вызывает агрегацию бактерий, подобную агглюцитации.
В обоих случаях сгусток фибрина, обволакивающий бактерии, создает подобие капсулы (псевдокапсулы), повышая устойчивость к фагоцитозу и бактерицидным факторам сыворотки.
Стафилолизины (гемолизины). Золотистый стафилококк продуцирует факторы, которые разрушают эритроциты, определяя один из характерных культуральных признаков S. aureus — прозрачную зону гемолиза вокруг колоний на кровяных средах (см. рис. 2). Они различаются по антигенным, физико-химическим свойствам, степени токсичности, лизируют эритроциты разных видов животных. Важно понимать, что эритроциты (и соответственно, гемолиз) — лишь удобный индикатор для выявления гемолизинов. На самом же деле они повреждают многие клетки, включая фагоциты. Этим объясняется их общая токсичность и, в частности, антифагоцитарная активность. Отсюда другие термины — стафилолизины или просто — токсины.
Дифференцируют четыре разновидности такого рода факторов: альфа-, бета-, гамма- и дельта-токсины/стафилолизины/гемолизины. Патогенетически наиболее важен альфа-токсин. Он связывается мембранными ганглиозидами, действуя как детергент, запускающий образование пор в зоне рецепции. Вслед за повышением проницаемости плазматической мембраны следует усиление трансмембранного потока ионов, которые (прежде всего Са2+) извращают внутриклеточный гомеостаз, приводя клетки к гибели. Спектр мишеней, поражаемых альфа-токсином, многообразен. Он повреждает лейкоциты, макрофаги, тромбоциты, тучные клетки, фибробласты, миоциты, гепатоциты, эндотелиоциты и др. При внутрикожном введении альфа-токсин вызывает некроз, а при внутривенных инъекциях оказывает токсическое действие на центральную нервную систему, миокард, паренхиматозные органы, сосуды, надпочечники и пр. Вместе с тем, результаты модельных экспериментов на животных нельзя безоговорочно переносить в клинику. Развитие стафилококковой инфекции часто не совпадает с динамикой антитоксического (в частности, альфа-антитоксического) иммунитета. Нередко выздоровление наступает вне всякой связи с накоплением антитоксических антител, а их высокий уровень в крови не гарантирует благополучного исхода патологического процесса. То же самое можно сказать о действии других гемолизинов: их значение в развитии стафилококковой болезни остается неясным.
Лейкоцидин (Panton—Valenine Leukocidin). Этот фактор продуцируется всего 2—3% штаммов S.aureus. Он действует как АДФ-рибозилаза на ферменты фосфолипидного метаболизма клеточных мембран, в том числе нейтрофилов (отсюда название). Это повышает проницаемость плазматической мембраны для Са2+, что ведет к каскаду вторичных эффектов, заканчивающихся гибелью клеток; в среде без Са2+ лейкоцидин не активен.
Редкое присутствие фактора объясняется следующим образом. Лейкоцидин относится к двухкомпонентным токсинам. Он состоит из двух неассоциированных белков, S и F (от англ. slow и fast — медленно и быстро элюируемые компоненты при ионообменной хроматографии), каждый из которых обладает собственной мишенью. Цитолитический эффект наблюдается только при их совместном действии. В штаммах, обладающих двумя токсинами (гамма-гемолизин и лейкоцидин), имеются три разных S-компонента (HlyA, HlyC и LukS-PV) и два F-компонента (HlуB и LukF-PV). От их сочетания зависит цитотоксическая активность цельного токсина. В частности, LukS-PV—LukF-PV — комбинация, которая не действует на эритроциты. Остальные сочетания дают три гамма-токсина — HlyA (HlуA—LukF-PV), HlyB (HlуA—HlуB) и HlyC (HlуC—HlyB). Они продуцируются практически всеми штаммами S. aureus, имеют высокую гемолитическую активность и слабее действуют на нейтрофилы.
Белок А. Пожалуй, это самый знаменитый фактор золотистого стафилококка, по крайней мере для специалистов, непосредственно не связанных с микробиологией. Он входит в состав клеточной стенки и обладает способностью вступать во взаимодействие с Fc-фрагментом IgG человека. Этот феномен, нашедший широкое применение в иммунологии, неспецифичен, так как реакция не затрагивает участков IgG, ответственных за распознавание антигенов. Если учесть, что Fc-фрагмент обеспечивает связывание IgG-опсонинов с фагоцитами и комплементом, то его блокада белком А может иметь негативные последствия для опсонофагоцитарной защиты.
Стафилокиназа. Растворение сгустков крови обеспечивает проникновение бактерий во внутреннюю среду. Так действует стафилокиназа. Переводя плазминоген в плазмин, она способствует разрушению фибрина, в том числе образуемого стафилококковой коагулазой. Это высвобождает инфицированные микротромбы, которые с током крови распространяются по организму. Этому содействует и повреждение экстрацеллюлярного матрикса соединительной ткани. В нем участвуют протеазы и гиалуронидаза стафилококка. Определенное значение имеет стафилококковая ДНК-аза. Ее подключение к инвазивности связано с расщеплением ДНК, накапливающейся в гнойном содержимом.
Лекарственная резистентность. Золотистый стафилококк печально знаменит своей быстрой адаптацией к антибиотикам и другим антибактериальным средствам. И хотя блестящий опыт первых испытаний пенициллина связан с излечением от стафилококкового сепсиса, сегодня это удается далеко не всегда. Количество резистентных стафилококков и спектр их устойчивости быстро росли на протяжении последних лет, и в настоящее время многие штаммы не чувствительны не только к классическому пенициллину, но и к антибиотикам последних поколений. Это эволюционно неизбежный процесс, отражающий экологическую пластичность бактерий. У стафилококков он связан с селекцией штаммов, разрушающих антибиотики. Главная роль принадлежит ферментам — плазмидзависимым бета-лактамазам. Они легко подстраиваются к новым пенициллинам и цефалоспоринам, а благодаря подвижности генов резистентности, входящих в состав R-транспозонов, быстро распространяются в микробных популяциях.
Лекарственная устойчивость — важнейший патогенетический фактор стафилококковых инвазий: затрудняя элиминацию возбудителя, он стабилизирует очаговые процессы, повышая вероятность осложнений. Кроме бета-лактамазоустойчивых пенициллинов (их история начинается с метициллина), цефалоспоринов и новых классов бета-лактамных антибиотиков (карбапенемы, монобактамы) для преодоления резистентности в состав препаратов предложено включать ингибиторы бета-лактамаз, например, сульбактам. Это повышает эффективность антибактериальной терапии, расширяя спектр чувствительных штаммов. Впрочем, и такой подход спасает не всегда. Большим злом являются метициллинрезистентные стафилококки, MRSA (от англ. methicillin-resistent S. aureus). Они устойчивы к бета-лактамным антибиотикам благодаря хромосомной добавке, кодирующей дополнительный пенициллинсвязывающий белок (РВР2а) — мутантную пептидогликановую транспептидазу. Несмотря на свое название (и в отличие от четырех главных транспептидаз), РВР2а слабо связывает бета-лактамные антибиотики, которые из-за этого лишаются антибактериальной активности. Такие штаммы резистентны к пенициллинам и цефалоспоринам (по крайней мере в реальных клинических дозировках), создавая серьезные трудности для этиотропной терапии. Приходится использовать другие антибиотики, в частности гликопептидного ряда. Препаратом выбора является ванкомицин, активный против MRSA. Однако недавно появились штаммы S. aureus, устойчивые к ванкомицину. Они создают тревожную перспективу, заставляя искать новые подходы к лечению инфекций, вызываемых множественно устойчивыми к лекарствам штаммами стафилококков.
Иммунитет
Золотистый стафилококк принадлежит к бактериям, которые вступают в напряженные иммунитетные взаимоотношения с человеком. В этом легко убедиться, если проанализировать содержание антител к различным (даже патогенетически инертным) антигенам S. aureus у практически здоровых людей. Их спектр и количество заметно превосходят аналогичные показатели для других бактерий, широко представленных в облигатной или факультативной микрофлоре. В этом отношении с ним сопоставимы лишь пиогенный стрептококк и пневмококк (см. «Стрептококк»). Иммунитет к золотистому стафилококку никогда не бывает полным. Любая травма, любое внедрение инородного тела создают почву для пиогенной инвазии. Случайная заноза вызовет нарыв как у ребенка, имевшего ограниченные контакты со стафилококком, так и у взрослого человека, обладающего богатым опытом таких столкновений. В этом экологическая сущность возбудителей оппортунистических инфекций: находясь в постоянном общении с человеком и иммунизируя его своими антигенами, они сохраняют потенциальную болезнетворность, реализуя ее в подходящих условиях (англ. opportunity — удобный случай).
Условия возникают не только извне (высокая инфицирующая дозировка, повреждение тканей, внедрение инородных тел), их формирует и сам инфицируемый организм. Речь идет о нарушениях гомеостаза, которые снижают резистентность к местным инвазиям и повышают вероятность системных осложнений. Сопротивление пиогенной инфекции во многом зависит от качества опсонофагоцитарных реакций. Их ослабление повышает чувствительность к стафилококку, способствуя стабилизации пиогенного процесса. Эндогенными факторами риска по развитию стафилококковых пиодермий являются диабет, гиперфункция сальных желез и изменение качества их секрета (в периоде полового созревания), возрастная эволюция апокринных потовых желез.
Стабилизация очагов пиогенной инфекции может быть связана с тем, что бактерии вызывают слабый иммунный ответ, который не обеспечивает образования необходимого количества опсонически активных антител. Установлено, например, что в отличие от стафилококковых поражений внутренних органов пиодермии не стимулируют синтеза антипептидогликановых антител — важного компонента опсонофагоцитарной защиты. У больных с хроническими стафилококковыми инвазиями (например, при остеомиелите) находили антитела, блокирующие опсоническую активность противостафилококковых антител здоровых людей. Кроме того, золотистый стафилококк способен скрываться от антител в сгустках фибрина и внутри самих фагоцитов. По той же причине иммунитет не срабатывает и при бактериемии. В любом случае, никто еще не сумел доказать, что содержание антител к каким-либо антигенам надежно коррелирует с устойчивостью против стафилококковых инвазий.
Много сил отдано иммунопрофилактике и иммунотерапии стафилококковых инфекций. На роль протективных антигенов претендовали разные продукты золотистого стафилококка. Наиболее широкие испытания прошел стафилококковый анатоксин. Его применение было нацелено на повышение устойчивости к S. aureus на основе антител против альфа-токсина. Опыты начались в 1930-х гг. под впечатлением блестящих результатов борьбы с дифтерией при помощи обезвреженного формалином токсина дифтерийной палочки. В нашей стране дело зашло так далеко, что препаратом стали широко иммунизировать беременных женщин для профилактики перинатальной гнойной патологии. Но накопилось столько противоречий, что плановая иммунопрофилактика была оставлена. Собственно этого следовало ожидать, исходя из патогенетической поливалентности золотистого стафилококка и скромной роли каждого из его факторов в развитии инфекционного процесса. Анатоксин (так же, как еще более загадочный препарат — антифагин) был рекомендован и для лечения хронических стафилококковых пиодермий. Механизм его действия в этом случае не ясен, но он, безусловно, не исчерпывается стимуляцией специфического иммунитета.
Иллюзорная идея о профилактической и терапевтической ценности анатоксина породила еще один препарат — противостафилококковый иммуноглобулин, широко разрекламированный как эффективное средство против септических стафилококковых инвазий. Его готовят из крови доноров, вакцинированных стафилококковым анатоксином, стандартизуют по антителам против альфа-токсина и именно их считают действующим началом. Но подобная логика не убеждает: вакцинация, не гарантирующая активного иммунитета, вряд ли способна обеспечить образование антител, эффективных в создании пассивного иммунитета. Введение иммуноглобулина (особенно больших дозировок при внутривенных инфузиях) может давать санирующий эффект не за счет нейтрализации бактериальных токсинов, а в результате подавления вторичной токсемии, связанной, в частности, с цитокинами. Синдром интоксикации (сложный сам по себе) — лишь часть патогенеза стафилококковой инвазии, и только с таких позиций следует обсуждать проблемы иммунотерапии стафилококковых инфекций.
Интоксикации
Наряду с пиогенными инфекциями, золотистый стафилококк способен вызывать специфические интоксикации, патогенез и клиническая картина которых целиком связаны с определенными бактериальными токсинами. Инвазия необязательна, хотя она может предшествовать или сопутствовать интоксикации. К специфическим токсинам золотистого стафилококка относятся эксфолиативный токсин, токсин синдрома токсического шока, энтеротоксин. В отличие от других патогенетически значимых факторов названные токсины кодируются плазмидами, умеренными бактериофагами или хромосомными генами, называемыми островами патогенности. Поэтому они продуцируются ограниченным числом стафилококковых штаммов, определяя уникальность их вирулентности.
Эксфолиативный синдром. Около 5% штаммов S. aureus образуют протеолитический токсин, разрушающий межклеточные контакты (десмосомы) зернистого слоя эпидермиса. Это ведет к отслоению (син. — десквамация, эксфолиация) поверхностных слоев эпидермиса и образованию изъязвляющихся (лопающихся) пузырей, наполненных серозным или гнойным содержимым. Инвазия минимальна, заболевание протекает по типу дерматита. Токсин носит название эксфолиатина, или эксфолиативного токсина. Он известен также как эпидермолитический токсин.
Эксфолиатин действует местно (т.е. в очаге инфекции) либо на расстоянии, вызывая системное поражение кожных покровов; в этом случае бактерии в зоне десквамации отсутствуют. Особенно драматично заболевание протекает у новорожденных и детей младшего возраста, когда общая интоксикация развивается на фоне незначительной, иногда оставленной без внимания пиодермии. Поражение начинается обычно с лица (вокруг рта), но уже через 48 ч захватывает значительную часть туловища и конечностей. После эритемы на коже возникают пузыри. Лопаясь, они обнажают глубокий болезненный слой эпидермиса. Температура, как правило, не повышена. Внешнее сходство с ожоговой поверхностью определило название — синдром ошпаренной кожи (син. — болезнь Риттера, пузырчатка новорожденных, генерализованный эксфолиативный дерматит, токсический эпидермолиз).
Картина выглядит устрашающей, но благодаря неинвазивному поражению эпидермиса и отсутствию у токсина других (кроме десмосом) мишеней все кончается благополучно: десквамация продолжается несколько дней и в течение двух недель пораженные участки заживают без рубца на фоне накопления в крови антител против эксфолиативного токсина. У детей более старшего возраста процесс ограничивается локальными поражениями — буллезное импетиго (лат. bulla — пузырь, impetus — внезапное нападение). Взрослые болеют редко.
Эксфолиатин (как и другие токсины S. aureus) — неоднороден. Описано две разновидности, одна из которых (тип А) контролируется хромосомными, а другая (тип В) — плазмидными генами. Они различаются по антигенным свойствам и нейтрализуются собственными антитоксинами, т.е. не создают перекрестного иммунитета. Еще одна микробиологическая деталь: по неразгаданной причине большинство штаммов, продуцирующих эксфолиатин, относятся к фагогруппе II (фаговар 71).
Пищевое отравление. Около половины штаммов золотистого стафилококка (чаще фагогруппы III) продуцируют токсины, которые, попадая в пищеварительный тракт, вызывают острейший гастроэнтерит с тяжелой рвотой и профузным поносом. Это наиболее распространенная пищевая интоксикация бактериальной природы: более половины этиологически расшифрованных пищевых отравлений в развитых странах связаны со стафилококком. Клиническая картина достигает пика через 2—3 ч после приема контаминированной пищи. Рвота отражает прямую стимуляцию энтеротоксинами окончаний блуждающего нерва, контролирующих рвотный рефлекс. Не исключено, что диарея также является следствием рефлекторного поражения центральной нервной системы, по крайней мере энтеротоксины плохо проникают через слизистые оболочки и всасываются в кровь. Впрочем, допускается возможность прямого поражения пищеварительного тракта. Кроме того, стафилококковые энтеротоксины обладают активностью суперантигенов, т.е. способны вызывать поликлональную (неспецифическую) стимуляцию Т-лимфоцитов за счет «незаконного» представления молекулами главного комплекса гистосовместимости. Они не процессируются внутри антигенпредставляющих клеток, а связываются с молекулами HLA, экспрессированными на их клеточной мембране. Одновременное вовлечение в реакцию множества клонов Т-лимфоцитов ведет к гиперсекреции цитокинов, вызывающих вторичную интоксикацию (см. «Болезнетворность бактерий»). Несмотря на тяжелые субъективные ощущения, симптомы бесследно проходят через несколько часов. Только самым маленьким детям и старикам иногда приходится вводить солевые растворы.
Стафилококковое пищевое отравление — пример чистой интоксикации: энтеротоксины накапливаются в пищевых продуктах, где размножаются энтеротоксигенные штаммы S. aureus, поступление бактерий в пищеварительный тракт (т.е. собственно инфекция) не имеет значения. В опытах на добровольцах доказано, что для взрослого человека патогенетически значимая доза энтеротоксинов составляет 1—4 мкг.
Известно 7 антигенных вариантов cтафилококкового энтеротоксина (SE) — SEА, SEВ, SEС1, SEC2, SEC3, SED, SEЕ. Вместе с пирогенными экзотоксинами Streptococcus pyogenes и стафилококковым токсином синдрома токсического шока (см. ниже) энтеротоксины объединены в группу суперантигенных токсинов. Их общими свойствами являются пирогенность и способность в 100000 раз и более повышать чувствительность кроликов к летальному эффекту эндотоксина.
Стафилококковые энтеротоксины неодинаковы по силе токсического эффекта, закодированы в генах умеренного бактериофага (SEA) или плазмидах (SEC, SED и SEE), обладают высокой термостабильностью (выдерживают кипячение, т.е. жесткий режим приготовления пищи) и устойчивостью к протеолитическим ферментам пищеварительного тракта. Другие токсины стафилококка в тех же условиях быстро разрушаются. Важное эпидемиологическое значение имеют широкое носительство золотистого стафилококка и его способность размножаться и продуцировать энтеротоксины в присутствии высоких (консервирующих) концентраций хлорида натрия.
Для обнаружения энтеротоксинов в пищевых продуктах предложены высокочувствительные тесты, но полезно помнить и о таком простом критерии, как общая обсемененность бактериями: пища, из 1 г которой высевается более 100 млн. золотистого стафилококка, может содержать опасную дозу токсина.
Синдром токсического шока (СТШ). Активное изучение этого заболевания началось в 1980 г., когда в США была зарегистрирована вспышка СТШ, впервые привлекшая серьезное внимание. СТШ характеризуется внезапным подъемом температуры, рвотой, профузным поносом, мышечными болями, быстро прогрессирующей гипотонией, вплоть до шока. Типичный признак — скарлатиноподобная сыпь, которая шелушится неделю и более, особенно на ладонях и стопах. Развивается почечная и респираторная недостаточность, изменения обнаруживаются в большинстве органов. Смертность — высокая.
В течение двух лет после первой вспышки в США была собрана информация почти о 1500 случаях СТШ. Вызывало удивление, что болели преимущественно молодые женщины, причем у большинства из них заболевание было связано с менструацией. Причину выяснили быстро. Оказалось, что все пострадавшие пользовались высокоадсорбционными вагинальными тампонами, которые менялись реже обычных тампонов, создавая благоприятные условия для размножения золотистого стафилококка и продукции его токсинов. Остроумно замечено, что тампонада влагалища, обильно контаминированного микробами, имитирует ситуацию, близкую к абсцессу. Скопление менструальной крови, детрита и пр. создает идеальную среду для размножения бактерий. И хотя в дальнейшем оказалось, что заболевание встречается при любой локализации стафилококковой инфекции, именно первая вспышка СТШ среди молодых женщин позволила разгадать его экологическую и патогенетическую сущность. Во всяком случае, настороженное отношение к качеству вагинальных тампонов и правилам их использования позволило избежать повторения крупных трагедий, сделав СТШ скорее поучительной историей, чем угрозой для будущего. По современной статистике, лишь треть от общего числа заболевших СТШ составляют менструирующие женщины.
При однократном обследовании вагинальное носительство золотистого стафилококка определяется примерно у 10% женщин, при многократном — у 60%. Возможно, что персистенции стафилококков способствует недостаток факторов, ингибирующих адгезию бактерий на вагинальном эпителии. Но главное то, что некоторые штаммы продуцируют очень сильный токсин, который, проникая путем трансцитоза через мукозальный эпителий, вызывает общее отравление организма — синдром токсического шока. TSST (от англ. toxic shock syndromе toxin) закодирован в умеренном фаге, но, к счастью, экспрессируется редко. Поэтому лишь немногие штаммы золотистого стафилококка способны вызывать СТШ. Подобно энтеротоксинам TSST обладает свойствами суперантигена, но их отношение к патогенетическому эффекту остается неясным. Мутанты TSST, практически лишенные суперантигенности, обладали таким же летальным действием для кроликов, как нативный субстрат.
Коагулазонегативные стафилококки (S. epidermidis, S. saprophyticus)
За последние пару десятилетий роль стафилококков в госпитальной патологии возросла за счет S. epidermidis и даже S. saprophyticus. Не обладая естественной инвазивностью, они получают свой шанс благодаря травматичным инструментальным процедурам, введению заместительных протезов и пр.
Эпидермальные стафилококки все чаще упоминаются среди возбудителей сепсиса, эндокардита (после кардиохирургических вмешательств), перитонита (больные на длительном перитонеальном диализе), воспалительных заболеваний мочевыводящей системы. Имеет значение способность S. epidermidis продуцировать особый полисахарид (межцеллюлярный полисахаридный адгезин), который обеспечивает прикрепление бактериальных агрегатов к полимерным материалам, в том числе к медицинскому инструментарию, протезам (например, клапанам сердца) и др. Это ведет к образованию биопленки, внутри которой чувствительность к антибиотикам гораздо меньше, чем у планктонных бактерий (см. «Болезнетворность бактерий»). Таким свойством обладают штаммы, выделяемые из клинического материала (крови), но не «сапрофитические» стафилококки, персистирующие на коже и слизистых оболочках здоровых людей. Признак регулируется группой генов, оформленных в виде оперона, и подчиняется фазовым (т.е. фенотипическим) вариациям, подвергаясь селекции под влиянием факторов внешней среды (например, при пассировании на кровяном агаре).
S. saprophyticus по неизвестным причинам вызывает острое воспаление мочевыводящей системы у здоровых людей. Чаще всего страдают женщины в возрасте 16—25 лет. Болезнь появляется вскоре после начала половой жизни и по симптомам напоминает инфекцию E. coli (см. «Кишечная палочка»). У многих больных отмечается пиелонефрит, который обычно заканчивается самоизлечением. Полагают, что стафилококки проникают в уринарный тракт по восходящему пути (с кожи рук и ног) и, обладая адгезинами к мукозальному эпителию, получают возможность для размножения. Их редко и в очень небольшом количестве обнаруживают в мочеиспускательном канале нормальных лиц обоих полов.
Внутрибольничная инфекция
Вероятность стафилококковых заболеваний возрастает для больных в стационарах, а также для рожениц и новорожденных в родильных домах. S. aureus принадлежит к числу важнейших возбудителей внутрибольничных инфекций, и именно здесь он обрел свою печальную известность. Недаром больницы называют меккой для вирулентных стафилококков.
Кроме высокой болезнетворности самого стафилококка следует помнить об общих факторах, способствующих развитию госпитальных инфекций:
1. Более широкое носительство потенциально опасных микроорганизмов среди больных и медицинского персонала. Это связано с повышением плотности контактов, предрасполагающих к интенсивному обмену микроорганизмами.
2. Формирование госпитальных эковаров (госпитальных штаммов) бактерий. Это происходит в результате селекции, связанной с постоянным воздействием негативных факторов, требующих от бактерий адаптивных перестроек (стерилизующие антисептики, лечебные антимикробные препараты, бактериостатические и бактерицидные факторы инфицированного организма). К фенотипическим особенностям госпитальных штаммов относятся резистентность к антибиотикам и другим антибактериальным препаратам, способность выживать в присутствии антисептиков, повышенная вирулентность (результат многократного пассирования через восприимчивый организм).
3. Наличие контингента с повышенной восприимчивостью к инфекции.
4. Появление дополнительных возможностей (входных ворот) для микробной инвазии: инфицирование ран после хирургических вмешательств, ожогов; применение диагностических и лечебных процедур, травмирующих кожу и слизистые оболочки (бронхоскопия, катетеризация, инъекции и пр.). На этом фоне опасными становятся даже безобидные бактерии, которые искусственно внедряются в организм, минуя непреодолимые для них барьеры, созданные эволюцией. Не случайно о заражении крови после врачебных процедур говорят как об инструментальном сепсисе.
Все сказанное имеет отношение не только к стафилококку, а к проблеме госпитальных инфекций в целом. Но именно S. aureus до сих пор остается одним из лидеров бактериального госпитализма. Спектр внутрибольничных стафилококковых инфекций повторяет бытовую патологию, но заболевания протекают тяжелее, а их вероятность (особенно раневых, хирургических инфекций) гораздо выше.
Из одной клетки, да не равны детки.
Стрептококки
- Особенности классификации.
- Взаимоотношения с человеком.
- Str. pyogenes (группа А): микробиологическая и патогенетическая многоликость.
- Str. agalactiae (группа В): перинатальная патология.
- Оральные стрептококки: норма и патология.
- Энтерококки: госпитальный оппортунизм.
- Анаэробные кокки: пептострептококки и пептококки.
Стрептококки относятся к роду Streptococcus семейства Streptococcaceae. Подобно стафилококку стрептококк — собирательное морфологическое понятие, которое объединяет грамположительные шаровидные бактерии (менее 2 мкм в диаметре), образующие цепочки (греч. streptos — скрученный, в виде цепи; рис. 1).

По другим (физиологическим) признакам стрептококки весьма неоднородны, что находит отражение и во взаимоотношениях их с человеком. Экологически стрептококки делятся на сапрофиты, которые участвуют в процессах молочнокислого брожения и находят промышленное (биотехнологическое) применение, и симбионты с широким спектром болезнетворности — от почти безвредных и даже полезных комменсалов человека и животных до возбудителей опасных заболеваний. Стрептококки принято относить к пиогенным коккам, что во многом связано с главным для медицины видом Str. pyogenes — мощным индуктором септических (гнойных) процессов. Однако пиогенность не исчерпывает болезнетворности стрептококков, а их лидер, Str. pyogenes, обладает рядом уникальных патогенетических потенций.
Стрептококки относятся к молочнокислым бактериям, т.е. обладают ферментативным (бродильным) типом метаболизма, среди конечных продуктов которого больше всего молочной кислоты. Фактически это анаэробы, которые получают энергию за счет брожения, но благодаря аэротолерантности хорошо размножаются в атмосфере кислорода. Родовой таксон Streptococcus включает несколько десятков видов, из которых около 20 имеют непосредственное отношение к человеку, входя в состав нормальной микрофлоры либо эпизодически паразитируя на слизистых оболочках. Стрептококки постоянно обитают в ротовой полости, носоглотке, толстом кишечнике, влагалище. Большинство из них обладают слабой болезнетворностью, и необходимы специальные условия для ее реализации. Пожалуй, единственный вид, Str. pyo-genes, по ряду признаков стоит ближе к патогенным бактериям. Он не просто опасен, а эпидемически опасен, вызывая не только спорадические, но и групповые заболевания.
Классификационные схемы стрептококков всегда учитывали экологические и патогенетические мотивы. Популярным остается деление стрептококков по способности лизировать эритроциты. В известном смысле это патогенетическая классификация, так как в ней учитывается способность бактерий к повреждению клеток — эритроцитов. Бета-стрептококки лизируют эритроциты, давая на кровяном агаре прозрачную зону полного гемолиза вокруг колоний (рис. 2). Альфа-стрептококки образуют зону зеленящего пигмента и поэтому называются зеленящими стрептококками. Строго говоря, это не гемолиз, а результат трансформации гемоглобина в метгемоглобин, подобно тому, что происходит в кровоподтеках (синяках). Гамма-стрептококки инертны в отношении эритроцитов и гемоглобина; они не меняют вид кровяного агара и называются негемолитическими.

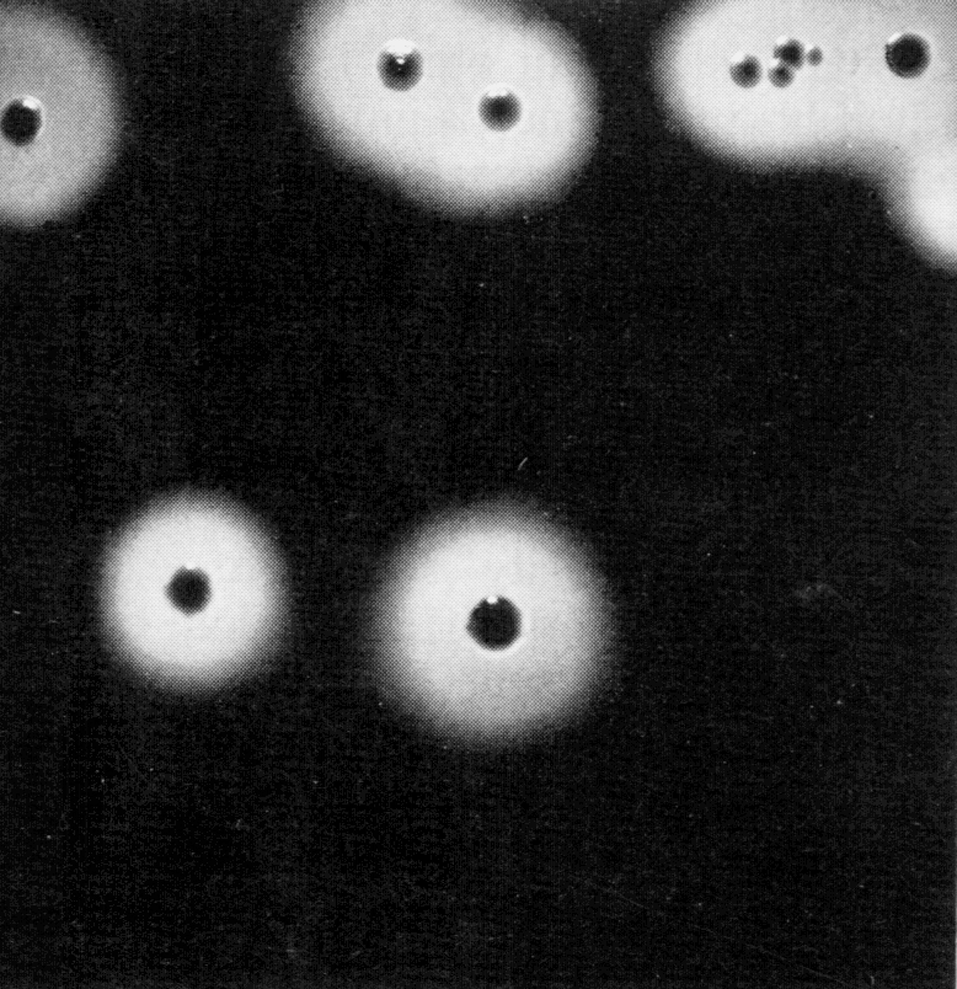
Большинство бета-гемолитических стрептококков человека являются болезнетворными. Они сравнительно редко выделяются от здоровых людей и всегда воспринимаются как потенциальная угроза для своего (обычно временного) хозяина и его окружения. Альфа-гемолитические стрептококки широко представлены в нормальной микрофлоре, особенно в ротовой полости (оральные стрептококки) и в толстом кишечнике (энтерококки; по современной классификации выделены в самостоятельный род Enterococcus). Это говорит о том, что альфа-гемолиз напрямую не связан с патогенностью, являясь скорее классификационным признаком, чем индикатором болезнетворности. Исключением является пневмококк (Str. pneumoniae) — один из важнейших патогенов человека. Он обладает мощной капсулой, которой лишены другие стрептококки, и именно она определяет его вирулентность. Негемолитические стрептококки входят в состав облигатной микрофлоры человека, но по своему видовому разнообразию уступают зеленящим стрептококкам. Они не играют роли в патологии человека или она минимальна.
Классификация стрептококков развивается в двух направлениях. Во-первых, было идентифицировано множество видов по биохимическим (ферментативным) признакам и факторам, претендующим на патогенность. Во-вторых, были разработаны методы серологической дифференциации стрептококков по групповым и типовым антигенам. Приоритет в этих исследованиях принадлежит американке Р.Ленсфилд, основополагающие работы которой (они относятся к тридцатым годам прошлого века) сыграли решающую роль в упорядочении знаний о стрептококках.
Деление на серологические (антигенные) группы за немногими исключениями основано на выявлении в реакции преципитации рамнозосодержащего полисахарида клеточной стенки, известного как С-субстанция. Преципитацией пользуются из-за того, что С-полисахарид часто экранирован другими компонентами клеточной стенки, и требуется экстракция, чтобы его обнаружить. В очищенном виде С-антиген является гаптеном, обретая иммуногенность в составе клеточных стенок. По его антигенным особенностям стрептококки разделены на 20 групп: A, B, C и т.д. Некоторые из них имеют статус вида, но есть и такие, которые включают несколько видов, дифференцируемых по другим признакам (например, группа С разделена на 4 вида). Принципиален факт: главный из медицинских стрептококков, Str. pyogenes, представляет единую серогруппу А, т.е. патогенетическая и серологическая классификации полностью совпадают. Известны стрептококки, которые лишены С-субстанции и классифицируются вне общей серологической схемы. К ним относится пневмококк и большинство зеленящих стрептококков, входящих в состав нормальной микрофлоры ротовой полости.
Бета-гемолитические стрептококки
Способностью лизировать эритроциты обладают стрептококки групп А, В (большинство штаммов), С, F и G. Это связано с продукцией гемолизинов, которые у стрептококка называются стрептолизинами. Дифференцируют два стрептолизина, S и О, которые различаются по чувствительности к кислороду: стрептолизин S (от англ. stable — устойчивый) не инактивируется в присутствии кислорода, стрептолизин О (от англ. oxygen-sensitive — чувствительный к кислороду) активен только в анаэробных условиях. Поэтому в обычных культурах (выращенных не в анаэростатах) гемолитическая активность обусловлена стрептолизином S. Впрочем, патогенетическая роль стрептолизина О, по-видимому, более значительна. Он обладает выраженной токсичностью для многих клеток (особенно для кардиомиоцитов), иммуногенен и антитела к нему (антистрептолизин О) служат индикатором активности стрептококкового процесса. Стрептолизин S менее токсичен и, не обладая иммуногенностью (отсутствие антител), не поддается специфической нейтрализации.
Большинство стрептококковых инфекций человека связаны с группой А, т.е. с Str. pyogenes, который в отличие от других бета-гемолитических стрептококков паразитирует только в организме человека. Гораздо реже сходные заболевания вызывают стрептококки В, С и G. Стрептококк группы В (Str. agalactiae) отличается своеобразной экологией — склонностью к паразитированию в родовом канале женщин; это делает его одним из важных факторов перинатальной патологии (см. ниже).
Str. pyogenes (группа А)
Антигены и факторы патогенности. Кроме С-группового полисахарида (группа А), в клеточной стенке Str. pyogenes содержатся еще несколько антигенов, по которым можно дифференцировать штаммы. Важнейшим из них является М-белок, структурно оформленный в виде филаментов, напоминающих пили/фимбрии грамотрицательных бактерий. По его антигенным особенностям идентифицировано более 80 М-типов (М-сероваров) пиогенного стрептококка, причем за редким исключением каждый из них содержит единственный вариант М-белка. М-белок (от англ. mucoid, так как колонии М-позитивных штаммов имеют мукоидную, т.е. слизистую консистенцию) представляет суперспирализованную молекулу, которая состоит из четырех участков (A—D), образующих филаментозные выросты, и двух фрагментов, которые заякоривают М-белок на мембране и в клеточной стенке (рис. 3). Эпитопная вариабельность связана с А-фрагментом, остальные участки более консервативны, что позволяет надеяться на создание универсальной противострептококковой вакцины.

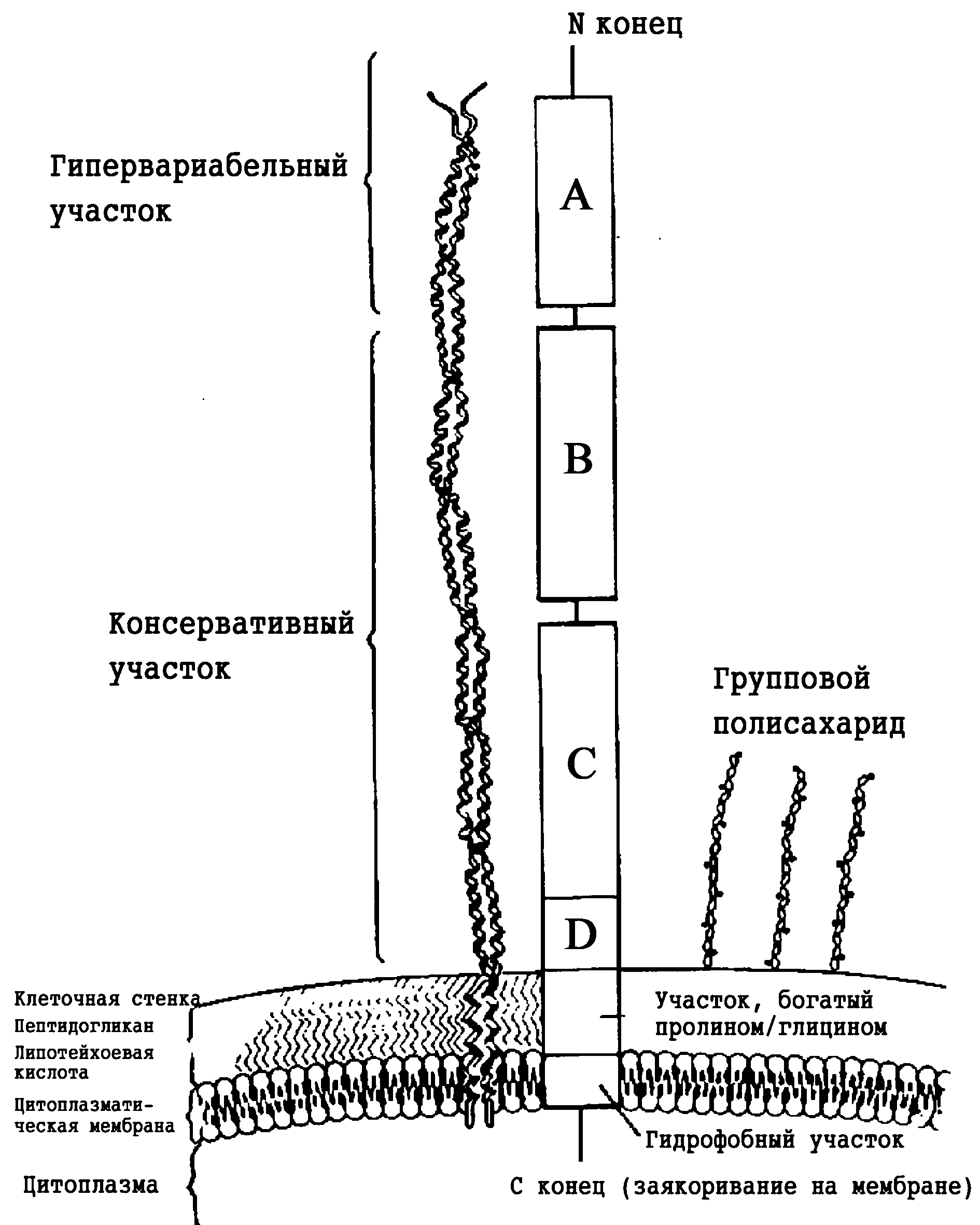
Типирование М-антигена оказалось полезным в эпидемиологических исследованиях, но главным явилось открытие его решающей роли в протективном иммунитете. Оказалось, что консервативные фрагменты М-антигена связывают фибриноген и Fc-участок IgG, формируя псевдокапсулу, экранирующую бактерии от фагоцитов. Антифагоцитарную активность М-белка дополняет фиксация сывороточного Н-фактора. Конкурируя с фактором В, он блокирует активацию альтернативного каскада комплемента, снижая эффективность антибактериальной реакции в опсонофагоцитарной системе. Снимая этот эффект, анти-М-антитела усиливают бактерицидность фагоцитов (нейтрофилов). М-белок имеет сайты связывания для фибронектина и CD46, способствуя адгезии стрептококка на эпителиальных клетках. Есть данные, что эта функция зависит от М-серотипа, отражая экологические варианты бактерий — респираторный и кожный стрептококки. Но и в этом случае не все ясно: некоторые высокоинвазивные и токсигенные (скарлатинозные) штаммы пиогенного стрептококка лишены М-подобных белков.
Узкая специфичность анти-М-антител определяет неудачную для человека иммунологию стрептококковых инфекций: иммунитет формируется раздельно для каждого из 80 М-сероваров Str. pyogenes. Кроме того, М-антигены обладают слабой иммуногенностью, и антитела к ним (по крайней мере с нейтрализующими свойствами) появляются с опозданием. Содержание М-белка снижается при культивировании бактерий на питательных средах, но быстро восстанавливается при пассировании на мышах. Это говорит о том, что стрептококки способны улавливать сигналы из внешней среды, контролируя образование фактора, играющего центральную роль в вирулентности. Установлено, что гены М-протеина (emm), входят в состав регулона, транскрипция которого находится под контролем позитивного регулятора mga, множественного генетического регулятора транскрипции стрептококка группы А. Экспрессия mga контролируется негативным регулятором Nra, который тормозит образование различных факторов патогенности стрептококка, в частности фибринонектинсвязывающего белка F2 и коллагенсвязывающего белка Сра. При ангине способность к продукции М-антигена падает по мере выздоровления, что лишний раз подчеркивает его отношение к агрессивности Str. pyogenes.
Кроме М-белка, к факторам антифагоцитарной защиты относятся стрептолизины, которые лизируют лейкоциты, подавляя фагоцитоз, и протеиназу, разрушающую С5а-фактор комплемента — мощный хемоаттрактант для нейтрофилов, способствующий их экстренной мобилизации в зону бактериальной инвазии. Ослабление этих механизмов снижает устойчивость к пиогенной инфекции, создавая предпосылки для опережающего размножения бактерий.
Кроме протеина М на поверхности многих штаммов имеются его рудименты — родственные и иммуноглобулинсвязывающие белки. Они кодируются теми же генами (emn или emn-related гены) и подобно М-белку связывают Fc-фрагмент IgG, имитируя защиту и, возможно, снижая опсоническую активность антител. Однако не все инвазивные и токсигенные штаммы располагают такими белками.
Большинство штаммов Str. pyogenes располагают капсулой, построенной из гиалуроновой кислоты. Так как гиалуроновая кислота входит в состав собственных тканей хозяина (это один из основных компонентов соединительной ткани), такая капсула лишена иммуногенности и не может быть атакована опсонинами. Теоретически это делает стрептококк неуязвимым, но он сам же лишает себя такого преимущества, продуцируя гиалуронидазу — фермент, разрушающий гиалуроновую кислоту. Поэтому патогенетическая (антифагоцитарная) роль капсулы сомнительна. Возможно, она способствует стабилизации инфекта в момент инвазии, но не имеет значения на последующих этапах инфекции. В частности, связываясь с гиалуронидатным рецептором CD44, капсула может быть одним из множества адгезинов, необходимых для закрепления стрептококка в верхних дыхательных путях.
Ненадежность собственной капсулы в известной мере компенсируется способностью Str. pyogenes фиксировать на своей поверхности белки хозяина (альфа-2-макроглобулин, фибриноген, плазминоген, фибронектин, витронектин, коллаген), создавая подобие капсулы.
В этой связи следует сказать и о липотейхоевой кислоте, пронизывающей клеточную стенку стрептококков. По инерции считают, что она играет едва ли не главную роль в закреплении (адгезии) бактерий на эпителии слизистых оболочек, связываясь с фибронектином, покрывающим эпителиоциты. Однако исследования с искусственными мутантами стрептококка заставляют усомниться в этом, выдвигая на первый план другие факторы адгезии (см. ниже).
Кроме антифагоцитарных факторов, инвазивность стрептококка определяется триадой ферментов, которые секретируются во внешнюю среду и разрушают тканевые барьеры на пути инфекции. К ним относятся стрептокиназа (фибринолизин), стрептодорназа (дезоксирибонуклеаза) и гиалуронидаза. Сходные факторы продуцируются и другими бактериями, но бета-гемолитические стрептококки наиболее активны в этом отношении. Не случайно перечисленные стрептококковые ферменты применяются в клинике для растворения внутрисосудистых тромбов (стрептокиназа), разжижения гнойных экссудатов и очищения инфицированных поверхностей (стрептодорназа), облегчения всасывания жидкостей, вводимых в ткани (гиалуронидаза). Именно таков механизм их патогенетического (инвазивного) действия в ходе естественного инфекционного процесса. Обратим внимание на природу фибринолитического эффекта стрептокиназы. В действительности, это собирательное название однотипных белков, которые не обладают способностью к прямому фибринолизу, но активируют плазминоген, переводя его в плазмин — протеолитический фермент, расщепляющий фибрин и другие белки, в частности С3-фактор комплемента — ключевой эффектор системы опсонической кооперации (фагоциты—комплемент—антитела) и, следовательно, антиинвазивной защиты. Стрептокиназный плазмин отличается важной особенностью: он не блокируется плазменными ингибиторами, обладая неконтролируемой активностью.
Важным свойством инвазивных стрептококков является способность фиксировать плазмин (плазминоген) на своей поверхности. Активируя металлопротеиназы и коллагеназы, плазмин способствует продвижению бактерий в тканях. Связывание плазмина или плазминогена (он трансформируется в плазмин стрептокиназой) происходит благодаря двум поверхностным ферментам, участвующим в синтезе макроэргических связей, глицеральдегид-3-фосфат дегидрогеназы и енолазы. Первый из них связывает ряд других тканевых белков (миозин, актин, фибронектин) и, по-видимому, играет роль в адгезивных реакциях стрептококка.
Много внимания уделяется стрептококковым модулинам. К ним относятся пирогенные экзотоксины со свойствами суперантигенов. Это более 10 белковых молекул (SpeA, SpeB, SpeC, SpeD, SpeF, SpeG, SpeH, SpeJ, стрептококковый суперантиген (SSA) и cтрептококковый митогенный экзотоксин Z (SMEZ, SMEZ-2), которые обладают свойствами неспецифических, или поликлональных, стимуляторов/митогенов для Т-лимфоцитов. Большинство из них закодировано в генах умеренных бактериофагов. Суперантигены одновременно связываются с молекулами НLA-2 (на поверхности антигенпредставляющих клеток) и выборочными фрагментами b-цепи Т-клеточных рецепторов (Vb), приводя к массированной продукции провоспалительных цитокинов. Полагают, что именно они вызывают многие эффекты, которыми отличается тяжелая стрептококковая интоксикация — cкарлатина и синдром токсического шока. При введении животным стрептококковые токсины вызывают пирогенный эффект и шок. Они значительно повышают чувствительность к эндотоксину, вовлекая его в патологический процесс при инвазивной стрептококковой инфекции. В этом отношении стрептококковые экзотоксины напоминают стафилококковые суперантигены, о которых говорилось выше (см. «Стафилококки»).
Примечательно, что SpeB оказался протеиназой из группы цистеиновых эндопептидаз. Благодаря широкому спектру активности она участвует не только в интоксикации организма (в частности, за счет образования активного ИЛ-1b из ИЛ-1b предшественника), но и в процессах колонизации и инвазии (расщепление фибронектина, витронектина, активация металлопротеиназы внеклеточного матрикса с повреждением эндотелия и др.). Во всяком случае нейтрализация токсина SpeВ (как и других токсинов) может предупредить тяжелые симптомы стрептококковой болезни.
Вместе с тем пирогенные экзотоксины продуцируют не только штаммы, выделяемые от больных с тяжелыми проявлениями стрептококковой инфекции. Токсины SpeА, SpeВ и SpeС могут секретировать культуры пиогенного стрептококка, получаемые от бессимптомных носителей, а также штаммы других, относительно безобидных (B—D) серогрупп. Полагают, что при прочих равных условиях причиной неодинаковой чувствительности к токсинам является разное количество антитоксических антител. Есть данные, что доля сывороток, не содержащих антитела к токсинам SpeА и SpeВ, в группе больных со стрептококковой инвазией в 2—3 раза выше, чем у здоровых доноров. Широким спектром нейтрализующей активности против стрептококковых суперантигенов обладает нормальный иммуноглобулин человека. Это связано с антитоксическими антителами: их удаление путем сорбции иммобилизованными стрептококковыми токсинами снижало нейтрализующий эффект. Увеличение в плазме уровня антител к токсинам после внутривенного введения иммуноглобулина подтверждено иммуноблоттингом.
Патология. Прикрепление к тканям хозяина — сложный процесс, в котором участвуют различные факторы Str. pyogenes: М-протеин, липотейхоевая кислота, комплекс фибронектинсвязывающих белков, капсула и др. Известно более 30 поверхностных компонентов, которые, взаимодействуя с белками сыворотки, секретов и соединительнотканного матрикса, словно ощупывают биотоп, куда попадают бактерии, координируя их дальнейшее поведение. Связывание с субстратом не является механическим феноменом. Оно рождает сигналы, которые определяют последовательность и интенсивность экспрессии генов, ответственных за реализацию болезнетворности стрептококка. Это ведет к развитию признаков вирулентного фенотипа, наиболее выгодных для конкурентной борьбы с хозяином. Подобная идеология, конечно, справедлива не только для стрептококка — это общий признак экологически зависимой детерминации микроорганизмов, которая распространяется и на взаимоотношения в системах «хозяин—паразит».
Общепризнано, что стрептококки не инвазируют клетки, хотя в инвитровых опытах этого удавалось добиться на основе фибронектинопосредованного взаимодействия с эпителиоцитами. Не исключено, что это один из механизмов, который обеспечивает персистенцию пиогенного стрептококка в фарингеальных эпителиоцитах, а также способствует пенетрации бактерий в субэпителиальную ткань.
Пиогенный стрептококк является классическим возбудителем пиогенных инвазий слизистых оболочек и кожи. Типовым выражением этого является флегмона (в зарубежной литературе термин «флегмона» заменен на «целлюлит» — лат. воспаление клетчатки) — острое (иногда острейшее) диффузное воспаление рыхлой соединительной ткани с тенденцией к распространению контактным путем и/или по лимфатическим протокам и межтканевым пространствам. В процесс (гнойное воспаление) вовлекаются лимфатические сосуды (лимфангиит) и регионарные лимфатические узлы (лимфаденит). В отличие от золотистого стафилококка абсцедирование наступает с опозданием и выражено умеренно. При внешнем разнообразии многие формы стрептококковой инфекции фактически представляют варианты флегмонозного воспаления. Так, ангину можно рассматривать как флегмону зева, рожу — флегмону кожи и т.д. Экссудат при стрептококковых флегмонах беден фагоцитами, так как лейкоциты, попадая в зону воспаления, разрушаются мощными бактериальными цитолизинами.
Поражает скорость фатальных исходов при стрептококковых инвазиях: при родовом сепсисе и пневмонии больные иногда погибают в течение считанных часов. История микробиологии оставила немало таких примеров, и именно страх перед сепсисом рожениц (родильная горячка) пробудил интерес к антисептике. К счастью, применение пенициллина, к которому Str. pyogenes и другие стрептококки (исключение — энтерококк) сохраняют до сих пор чувствительность, резко снизило угрозу стрептококковых инфекций. Сегодня их удельный вес в общей структуре пиогенных осложнений сравнительно невелик, но и того, что осталось, вполне достаточно, чтобы с «уважением» относиться к этому возбудителю, памятуя, что история повторяется, если ей пренебрегать.
Пиогенная инвазия не исчерпывает всего спектра стрептококковой патологии. Str. pyogenes угрожает тяжелой общей интоксикацией — высокая температура, головная боль, жажда, ослабление аппетита, возбуждение, тахикардия, аритмия, рвота. Ее грозными проявлениями служат синдром токсического шока и его тонзиллярная форма — скарлатина. Полный симптомокомплекс сегодня встречается редко, так как стрептококковые инфекции хорошо лечатся антибиотиками. Но, как уже говорилось, история оставила суровую память о тяжелых, нередко смертельных последствиях банальной ангины, стрептококковой родильной горячки, менингита и пр.
Наконец, для Str. pyogenes характерны отсроченные осложнения — ревматизм и гломерулонефрит, протекающие совершенно иначе, чем события в первичном очаге инфекции. Они рассматриваются как результат уникальной антигенной агрессии, влекущей за собой классические образцы микробзависимой иммунопатологии.
Ангина. Ангиной (лат. angere — сжимать, душить, давить) называется острое воспаление зева (фарингит/тонзиллит), которое равномерно захватывает все его части (дужки зева, мягкое небо, миндалины и среднюю часть глотки) или ограничивается одной из них, чаще всего небными миндалинами (тонзиллит). Фарингит сопутствует многим вирусным (более 80% всех катаральных ангин) и бактериальным инфекциям, но пиогенная инвазия зева типична прежде всего для стрептококка. Более 90% cептических (гнойных) ангин связано с Str. pyogenes, и именно ангина является самой распространенной стрептококковой инфекцией, с которой начинаются ее осложнения. Процесс может ограничиться катаральными проявлениями (это характерно для детей младшего возраста), но в типичных случаях стрептококковая ангина протекает как гнойное воспаление слизистой оболочки зева с яркой гиперемией, отеком, увеличением миндалин и общей интоксикацией. Из местных осложнений обычным является воспаление регионарных лимфатических узлов (шейный лимфаденит). Возможно развитие отита, синуситов, поражение более глубоких отделов респираторного тракта. Первичный и каждый из вторичных очагов инфекции может стать источником бактериемии, угрожая септической генерализацией (менингит, эндокардит и пр.).
Респираторные стрептококки отличаются от кожных по М-антигенам. Ангину вызывают М-типы 1, 3, 5, 6, 14, 18, 19 и 24, тогда как пиодермальные стрептококки чаще принадлежат к типам 2, 49, 57, 59, 60 и 61. Болезнь не всегда оканчивается после завершения местного процесса. Для Str. pyogenes характерны апиогенные осложнения, которые проявляются спустя 1—4 нед после острой инфекции. К ним относятся ревматизм и гломерулонефрит, причем поражению почек чаще предшествуют стрептококковые инфекции кожи, а ревматизм обычно возникает после ангины. Ревматизм и гломерулонефрит рассматриваются как иммунопатологические процессы, которые развиваются в организме, сенсибилизированном к антигенам стрептококка. Поэтому аллергическая перестройка, наряду с инвазивным и токсическим компонентами, считается важным фактором патогенеза стрептококковых инфекций. Ангину могут вызывать стрептококки групп С и G. Но ревматогенная сенсибилизация встречается лишь в случае пиогенного стрептококка. Это требует дифференциальной диагностики ангин для профилактики ревматизма, которая проводится путем длительного лечения антибиотиками. Для этого требуется экспресс-индикация антигенов группы А в очаге инфекции, подтвержденная выделением чистой культуры стрептококка.
Гломерулонефрит связан с отложением в почечных капиллярах (гломерулах) комплексов антиген-антитело. Природа стрептококковых антигенов, ответственных за формирование патогенетически значимых иммунных комплексов, неизвестна, но по крайней мере один из них связан с плазматической мембраной бактерий. Стрептококковые антигены играют роль наводящего фактора, способствуя избирательной фиксации иммунных агрегатов на базальной мембране гломерул. Такую функцию могут выполнять многие компоненты бактерий, в частности плазминсвязывающий белок, нефритогенные стрептокиназы, М-белки. Это инициирует развитие иммунокомплексной патологии — острой воспалительной реакции, которая сопровождается повреждением почечных капилляров. Большинство случаев заканчивается полным выздоровлением через одну—две недели, но бывают и тяжелые осложнения. Серьезную опасность представляет хронизация процесса с развитием почечной недостаточности.
Установлена связь гломерулонефрита с определенными М-типами пиогенного стрептококка. При кожных инфекциях нефритогенные штаммы обычно относятся к М49, М57, М59, М60; при ангинах — к М1 и М12.
Ревматизм до открытия антибиотиков развивался после стрептококковой ангины в 0,1 — 3,0% случаев, обычно у детей и лиц юного возраста. Ревматические осложнения чаще наблюдаются при тяжелой ангине, хотя могут возникать и при легких (порой оставшихся незамеченными) формах. Первое проявление ревматизма обычно сопряжено с острым поражением суставов (асептический полиартрит), но за этим следует главная патология, панкардит — воспаление всех слоев сердца (эндокарда, миокарда, перикарда). Наиболее коварен эндокардит. Трансформируясь в хронический воспалительный процесс, он деформирует клапаны сердца, нарушая их функции. Заболевание начинается с небольших (субклинических) повреждений, которые возбуждают реактивное разрастание фиброзной ткани, и именно этот, репаративный по своей сути процесс ведет к необратимой патологии — стенозу и недостаточности клапанов, обычно митрального и аортального. По аналогичной схеме (острое экссудативное воспаление — хроническое гранулематозное воспаление — фиброз) развиваются миокардит и перикардит. Это ведет к функциональной недостаточности сердечной мышцы, которая при перикардите усугубляется ослаблением сердечных сокращений из-за фиброзного перерождения фибрина на поверхности перикарда (панцирное сердце). Поражение сердца наблюдается примерно у половины больных с ревматическим артритом, но иногда возникает без клинических признаков острого ревматизма. В таких случаях процесс тянется годами, и порок сердца проявляется неожиданно для больного, так как формально не связан с предшествующими заболеваниями. Об отношении к стрептококку говорят высокие титры противострептококковых антител, в частности анти-О-стрептолизина.
Несмотря на обилие гипотез, патогенез ревматизма остается загадкой. Доминирует иммунопатогенетическая концепция, согласно которой стрептококк индуцирует гиперреактивность (аллергию) к собственным антигенам и, возможно, к антигенам (аутоантигенам) хозяина. Str. pyogenes содержит по крайней мере 4 антигена, которые перекрестно реагируют с тканью сердца и способны стимулировать образование аутоантител и Т-лимфоцитов, сенсибилизированных против кардиальных антигенов. Не исключено, что это запускает и поддерживает аутоагрессию, которая наслаивается на другие патогенетические механизмы. Впрочем, роль аутоиммунных реакций при ревматизме окончательно не доказана, эта версия встречает ряд возражений, впрочем, как и другие концепции ревматического процесса.
Синдром токсического шока. С конца 80-х гг. во многих цивилизованных странах наблюдается рост инвазивной стрептококковой инфекции. Ей сопутствует быстрое развитие тяжелой формы интоксикации, подобной стафилококковому синдрому токсического шока. Болеют взрослые люди, особенно после 65 лет. Заболевание не передается окружающим, хотя лица, контактирующие с больным, могут стать носителями потенциально опасного микроба. Большинство случаев связано с М-типами 1 и 3, смертность составляет 30—60%. Входными воротами являются кожа, слизистая оболочка влагалища или матки. Признаками (не считая местных симптомов) служит снижение кровяного давления (< 90 мм рт.ст.) и множественная органная недостаточность. Можно думать, что именно от такой болезни погиб тургеневский Д. Базаров из «Отцов и детей». Он заразился при вскрытии женщины, погибшей от родильной горячки.
В патогенезе токсического шока участвуют пирогенные токсины SpeА, SpeВ и SpeС, а также ряд новых суперантигенов стрептококка. Опыты с удалением из организма больных Т-клеток, чувствительных к пирогенным токсинам, показывают, что с ними связаны по крайней мере некоторые проявления стрептококковой интоксикации. Об этом же говорят положительные результаты лечения больных внутривенными инъекциями больших дозировок нормального иммуноглобулина. Эффект связывают с наличием нейтрализующих антител против экзотоксинов SpeА, SpeВ и SpeС. Именно их недостаток может быть фактором, предрасполагающим к заболеванию.
Скарлатина представляет собой разновидность пирогенной интоксикации, сопутствующей стрептококковой ангине. Ее отличительный признак — яркая (англ. scarlet — алый) мелкоточечная сыпь, которая сливается в общую эритему и заканчивается шелушением. В остальном течение не отличается от обычной ангины с ее возможными осложнениями и прогнозом. Болеют в основном дети 5—10 лет. Подобно стрептококковой ангине заболевание отличается контагиозностью, формируя очаги эпидемических вспышек. Эволюция инфекции часто не предсказуема. Она может переходить в тяжелейшие токсические формы, напоминающие синдром токсического шока. Не случайно стрептококк заслужил репутацию возрождающегося возбудителя опасных инфекций.
Скарлатинозные стрептококки продуцируют пирогенные экзотоксины, которые действуют на стенку сосудов, вызывая сыпь (эритему). Из большой группы пирогенных экзотоксинов в развитии скарлатины наиболее значительную роль играют три серологических варианта — SpeА, SpeВ и SpeС. Способность к их образованию зависит от умеренных фагов и может передаваться от токсигенных нетоксигенным штаммам. Фаги, содержащие tox+-гены, циркулируют только среди штаммов Str. pyogenes. Другие стрептококки не инфицируются такими фагами и не вызывают скарлатину. Токсины SpeА, SpeВ и SpeС связаны общими эпитопами, что определяет возможность перекрестного иммунитета, оставляя небольшую вероятность повторных заболеваний. По сути, это те же токсины, известные для многих штаммов стрептококка группы А и даже других серогрупп. Поэтому не ясно, почему скарлатину вызывают лишь некоторые штаммы группы А. Стрептококку не подходит концепция о клонотипах: генетически не отличимые штаммы способны вызывать (или не вызывать) заболевания различной тяжести с разным спектром клинических проявлений.
Механизм действия пирогенных токсинов включает элементы опосредованной (вторичной) токсичности. Сегодня известно, что речь идет о суперантигенах, способных к поликлональной, или неспецифической (т.е. антигеннезависимой) стимуляции Т-лимфоцитов. Это сопровождается гиперпродукцией цитокинов с мощным патогенетическим потенциалом. Может иметь значение и то, что скарлатинозные токсины повышают чувствительность к О-стрептолизину и липополисахаридному эндотоксину грамотрицательных бактерий. Наконец, есть сообщения о прямом повреждающем действии пирогенных токсинов на эндотелиальные клетки. Это способствует массивной утечке плазмы и гипотензии, характерной для синдрома токсического шока.
Важным качеством скарлатинозных штаммов является эпидемичность, т.е. способность вызывать групповые заболевания, особенно в детских коллективах. Впрочем, этот признак не уникален, так как эпидемические вспышки случаются и при обычных стрептококковых ангинах. Тем не менее именно скарлатина принадлежит к наиболее контагиозным стрептококковым инфекциям.
Интересно обстоит дело с иммунитетом против скарлатины. Антитела к пирогенным токсинам нейтрализуют их активность, предупреждая рецидивы сыпи, но не влияют на устойчивость к стрептококковой инвазии, защита от которой, как и при других инфекциях, зависит от анти-М-антител. Поэтому лица, перенесшие скарлатину, обычно не болеют ей повторно, но восприимчивы к новым для себя М-типам стрептококка. В таких случаях возникает только ангина, а больные являются завуалированным источником «скарлатинозных штаммов». К счастью, в настоящее время случаи скарлатины единичны.
Поражения кожи. Неповрежденная кожа непроницаема для стрептококков. Но даже малейшие, незаметные для глаза микротравмы становятся входными воротами для пиогенной инфекции кожи и соседних тканей.
Поражение поверхностных слоев кожи носит название стрептококковой пиодермии или контагиозного импетиго (лат. impetus — натиск). Мы говорили о нем в очерке о стафилококках, так как импетиго имеет двойную этиологию (Str. pyogenes, S. aureus) и нередко развивается как смешанная инфекция. В таких случаях процесс начинает стрептококк, а стафилококк присоединяется как вторичная инфекция. Некоторые авторы вообще сомневаются в патогенетической значимости стафилококка. Подобно ангинам, импетиго изредка вызывается стрептококками групп В, С и G.
Инфицируется эпидермис, и поначалу очаги поражения выглядят как везикулезные высыпания, напоминая ветряную оспу. Но они быстро трансформируются в пустулы (т.е. обретают гнойный характер), которые изъязвляются, покрываясь корочкой лимонного цвета, из-под которой легко выделить стрептококк или смешанную культуру стрептококка и стафилококка. Инфекция (ее развитию способствует жаркий, влажный климат) чаще поражает детей (обычно летом) и может протекать по типу вспышек, передаваясь контактным путем. Внедрению бактерий способствуют расчесывания, укусы насекомых. Ретроспективно этиологический диагноз можно подтвердить по нарастанию антител против антигенов стрептококка, хотя профиль иммунного ответа несколько отличается от реакции при ангинах (в частности, регистрируется слабое нарастание антител против О-стрептолизина). Отмечены и особенности штаммов, вызывающих поражения кожи и респираторного тракта. При импетиго чаще выделяются М-типы с высокими номерами (М49, 57—61), которые, по-видимому, обладают способностью к более длительному выживанию на коже.
Само по себе заболевание неопасно, но, как и при любой стрептококковой инфекции, вероятны осложнения. Главным из них является гломерулонефрит, который развивается у 1—10% больных через две-три недели после перенесенной инфекции. Это больше, чем при ангинах и, возможно, связано с тем, что при импетиго чаще встречаются нефритогенные штаммы пиогенного стрептококка.
Рожа. Str. pyogenes — единственный возбудитель острейшего флегмоноподобного воспаления кожи, давно известного в России под названием «рожа». Термин эмоционально отражает облик больного при стрептококковых поражениях лица. В зарубежной литературе заболевание носит название erysipelas (греч. красная кожа). Инфекция распространяется в субэпителиальной ткани лица, головы, нижних конечностей, реже — других частей тела. Возбудитель внедряется через ничтожные повреждения кожи, хотя допускается и вероятность гематогенного распространения, например из очагов стрептококковой ангины. Признаки воспаления (покраснение, отечность) при роже выражены чрезвычайно ярко, зона поражения резко отграничена от здоровой ткани. Экссудат имеет серозно-фибринозный характер, нейтрофилы присутствуют в небольшом количестве. Выделить возбудителя из центральной зоны поражения не удается, но изобилие стрептококков легко обнаружить в пунктатах внешне здоровой кожи на расстоянии примерно 3 см от демаркационной линии воспаления. Здесь стрептококк заполняет лимфатические сосуды, и приходится лишь удивляться, почему столь мощное скопление пиогенных бактерий не сопровождается воспалительной реакцией. Но так или иначе, именно на этом рубеже срабатывает барьер, препятствующий дальнейшему распространению инфекции.
Особенности рожистых штаммов не известны. Они располагают пирогенными токсинами, типичными для Str. pyogenes, однако их значение не подтвердилось: вакцинация «скарлатинозным» токсином не дает профилактического эффекта, а антитоксическая сыворотка не влияет на развитие рожистого воспаления. Патогенетическая неопределенность рожи вынуждает мириться с расплывчатой декларацией о том, что многое здесь зависит от реактивности (точнее, гиперреактивности) организма, т.е. от своеобразия взаимоотношений в индивидуальных системах «макроорганизм—микроорганизм». Действительно, есть люди, которые подвержены рецидивам заболевания, тогда как большинство, заражаясь теми же штаммами, устойчивы к рожистому воспалению. Поэтому случаи рожи всегда единичны, групповые заболевания не известны. Главное осложнение — переход в типичную флегмону, когда подкожная клетчатка и мышечная ткань подвергаются гнойному пропитыванию, создавая опасность генерализации инфекции.
Раневые инфекции. Входными воротами являются открытые раны от травм или хирургических вмешательств, создающие условия для внедрения массивных дозировок бактерий. Нередко наблюдается смешанная инфекция, например с бактероидами. В наиболее агрессивной форме процесс (его называют госпитальной гангреной или некротизирующим фасциитом) захватывает подкожную клетчатку и мышечную ткань. Реальной угрозой служат септическая бактериемия и шоковый токсический синдром, историческим примером которого является уже не раз упоминавшийся родовой (пуэрперальный) сепсис, связанный с внесением пиогенного стрептококка в травмированные ткани родовых путей и матки при акушерских процедурах. Смертность при стрептококковых инвазиях достигает 20—50%, несмотря на чувствительность возбудителя к пенициллину и другим антибиотикам. Попытки создать вакцину не увенчались успехом. Сегодня на фоне почти полного исчезновения стрептококковых инвазий такая необходимость кажется излишней. Современные клиники с хорошо налаженным асептическим режимом скорее всего исключают такую потребность.
Иммунитет. Str. pyogenes вступает в весьма напряженные иммунологические взаимоотношения с человеком. В крови даже здоровых людей содержатся антитела к многочисленным структурным и секреторным продуктам стрептококка (рис. 5).

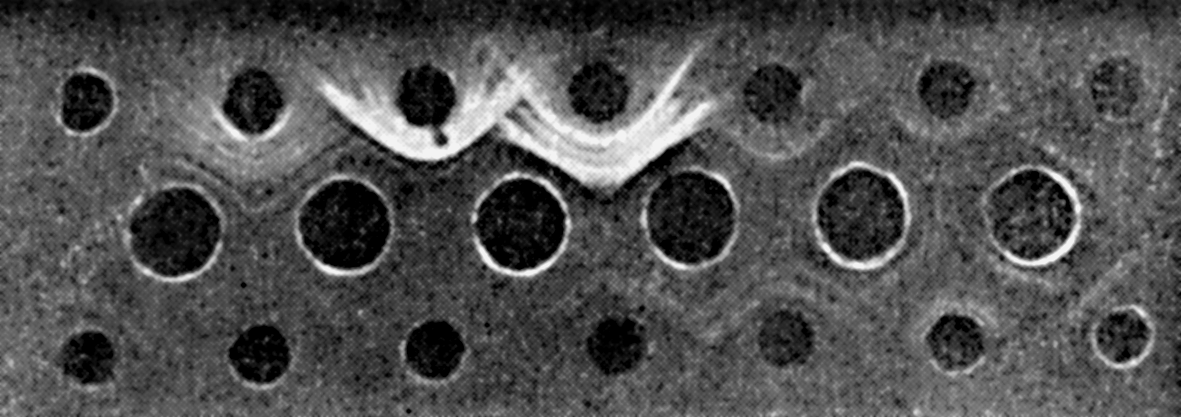
Их содержание возрастает после перенесенных стрептококковых инфекций, что служит диагностическим критерием. Особенно популярен тест на антитела к О-стрептолизину, хотя с аналогичной целью можно использовать и другие показатели (антигиалуронидаза, антистрептокиназа и пр.). Парадокс в том, что даже высокие титры этих антител не говорят о невосприимчивости к стрептококку. Единственное исключение — антитела против пирогенных токсинов, которые защищают от рецидивов скарлатины. Впрочем, как уже говорилось, это то правило, которое подтверждает исключение: способность специфически нейтрализовать токсины не предохраняет от пиогенной инвазии, а лишь предупреждает развитие одного из ее симптомов — скарлатинозной интоксикации.
Иммунитет против стрептококковых инвазий связан с антителами против М-антигенов, которые в содружестве с комплементом усиливают бактерицидные реакции нейтрофилов. Гетерогенность М-белка исключает приобретение надежного иммунитета, предохраняя лишь против реинфицирования штаммами идентичного М-типа. Необычный характер стрептококковых осложнений, их возрастные и индивидуальные особенности до сих пор рождают споры. Акцент традиционно делается на иммунопатогенез, т.е. на поражения, опосредованные иммунологическими механизмами. В этой связи обсуждается роль аллергии, иммунокомплексной патологии, аутоагрессии, суперантигенности стрептококковых токсинов и М-белка. Судьба этих гипотез остается неопределенной.
Str. agalactiae (группа В)
Этот стрептококк давно известен в ветеринарии, являясь одним из возбудителей мастита у коров. Отсюда и видовой эпитет — agalactiae, отражающий главный симптом заболевания — отсутствие молока у больных животных. Значение Str. agalactiae для медицины определяется его своеобразной экологией: он охотно колонизирует слизистую оболочку влагалища. Str. agalactiae выделяется от 10% здоровых женщин, а при беременности этот показатель возрастает до 25%, поэтому наибольшую опасность Str. agalactiae представляет для новорожденных, которые заражаются, проходя через инфицированный родовой канал. Примерно у 1 из 100 новорожденных инфекция имеет клинические последствия, проявляясь в первые 24 ч после рождения. Факторами риска являются недоношенность, преждевременный разрыв околоплодных оболочек, высокая обсемененность родовых путей и отсутствие специфических антител у матери. Местом первичной колонизации и патологии у ребенка служат легкие, но возбудитель может попасть в кровь, создавая опасность септических осложнений (менингит). При менингите летальность достигает 50% (по некоторым данным 100%), и даже экстренное назначение антибиотиков часто не спасает больного. Заражение новорожденных может быть и результатом госпитального или бытового инфицирования. В таких случаях развивается поздний (отсроченный) вариант инфекции, который проявляется обычно через 7—10 дней, иногда много позже. Он протекает менее остро и клинически не столь определенно, хотя опасность генерализации (менингит) сохраняется.
Полагают, что вирулентность связана с полисахаридной капсулой. В отличие от гиалуронидатной капсулы стрептококка группы А она состоит из «чужих» компонентов и поэтому обладает иммуногенностью. Известно восемь серотипов В-капсулы. Более половины неонатальных инфекций связаны с типом III. Ее вирулентность определяется присутствием сиаловой кислоты, которая создает инертность по отношению к альтернативному пути активации комплемента. Антикапсульные антитела опсонизируют бактерии и, привлекая классический механизм комплемента, защищают от инвазии. Это означает, что для профилактики неонатальной болезни лучше всего были бы материнские IgG-антитела против данного серотипа. Проходя через плаценту, они служили бы надежной защитой от инфекции. Но, будучи Т-независимым антигеном, капсульный полисахарид является слабым стимулом для образования IgG-антител и не может быть сам по себе использован для создания эффективной вакцины. Для этого требуются конъюгированные полисахарид-протеиновые вакцины, подобные вакцине из полисахарида b Haemophilus influenzae. Такие препараты уже появились и в настоящее время проходят испытания.
У взрослых стрептококки группы В изредка вызывают целлюлит (флегмону) и его генерализованные осложнения — артриты и менингит. Группу риска составляют пожилые, диабетики и алкоголики.
Оральные стрептококки
Имеется около десятка видов стрептококка, которые входят в состав нормальной микрофлоры ротовой полости и верхних дыхательных путей человека. Они персистируют на эпителии слизистой оболочки щек, носоглотки, в десневых карманах, на поверхности зубов. В своем большинстве это альфа-гемолитические (зеленящие) стрептококки. Они вносят определенный вклад в формирование и стабилизацию микробиоценоза соответствующих биотопов, участвуя в формировании колонизационной резистентности слизистых оболочек.
Подострый септический эндокардит (endocarditis lenta). Не обладая инвазивностью, зеленящие стрептококки активно не выходят за пределы своих естественных биотопов и в этом смысле не представляют опасности. Но экология ротовой полости такова, что в ней постоянно возникают возможности для пассивного внедрения инфекта. Микротравмы возникают даже при обычном жевании, чистке зубов, а такие процедуры, как экстракция зуба, тонзиллэктомия, заметно повышают вероятность заноса бактерий ротовой полости в кровяное русло. Установлено, например, что после удаления зуба оральные стрептококки попадают в кровь в 30% случаев.
Бактериемия после продолжительного жевания, по-видимому, обычное явление. Она носит транзиторный характер и не имеет последствий, так как стрептококки и другие бактерии элиминируются системой внутрисосудистого клиренса крови. Но исход может быть иным, если бактерии, находя участки для закрепления в кровяном русле, получают шанс для размножения.Стрептококки не поражают нормальный эндотелий, но при повреждениях его резистентность к микробной колонизации падает. Главной мишенью служат клапаны сердца, измененные ревматическим воспалением, атеросклерозом, врожденными пороками. Они (чаще аортальный и митральный клапаны) подвергаются изъязвлению, пораженная поверхность покрывается полипозными тромбами, внутри которых бактерии защищены от бактерицидных факторов крови. Распадаясь, тромбы создают опасность эмболических поражений почек, головного мозга, селезенки. У больных держится субфебрильная температура с периодическими подъемами. Хроническая интоксикация ведет к анемии, системному васкулиту, дистрофии паренхиматозных органов. Процесс заселения напоминает биопленку, в формировании которой принимают участие реактивные системы хозяина. Без лечения болезнь тянется месяцами и заканчивается смертью. Антибактериальная терапия может остановить септический процесс, но не развитие порока сердца (результат реактивного склероза).
Большинство случаев септического подострого эндокардита обусловлено Str. mitis, зеленящим стрептококком, лишенным группового антигена. В 5—10% случаев заболевание вызывают энтерококки. Длительный, вялый характер инфекции отражает слабую вирулентность возбудителей, реализация которой зависит от стечения «случайных обстоятельств». Этих обстоятельств должно быть гораздо больше, чем при остром эндокардите, связанном с пиогенным стрептококком и другими агрессивными бактериями. В таких случаях процесс завершается быстрой деструкцией клапанов и летальным исходом в течение нескольких дней или недель.
Кариес. Оральные стрептококки сопряжены с одним из самых распространенных заболеваний ротовой полости — кариесом. Процесс начинается с повреждения зубной эмали, а затем переходит на более глубокие слои зубной ткани. Разрушение происходит на фоне формирования биопленки (см. «Болезнетворность бактерий»), в результате образования зубного налета, или бляшек. В состав бляшек входят многие бактерии, но главным инициатором является Str. mutans. Он обладает двумя свойствами, которые оказывают решающее влияние на развитие процесса. Во-первых, Str. mutans в изобилии продуцирует леван — полисахарид, обеспечивающий прикрепление бактерий к зубной поверхности, а во-вторых, подобно всем стрептококкам он образует молочную кислоту из углеводов, которая растворяет зубную эмаль. Хозяин не реагирует на начало процесса и не противодействует его развитию. Воспалительная реакция возникает лишь на поздних стадиях, когда через поврежденную эмаль бактерии проникают в пульпу зуба.
Энтерококки
Бактерии этой группы включают несколько видов, объединенных своеобразной экологией (входят в состав нормальной микрофлоры толстого кишечника человека и теплокровных животных), общностью антигенной структуры (содержат групповой антиген D), высокой устойчивостью к факторам внешней среды (рост в присутствии 10% желчи, 5% хлористого натрия) и необычной для стрептококков резистентностью к антибиотикам. Своеобразие энтерококков явилось поводом для их выделения в самостоятельный род — Enterococcus. К патологии человека имеют отношение два вида — E.faicium и E.faecalis.
Экологические особенности энтерококков имеют патогенетические параллели. Безвредные в естественной для себя среде обитания (кишечник), энтерококки способны вызывать и поддерживать воспалительные процессы в соседних участках тела — в мочевыводящей системе и брюшной полости. Они попадают сюда при травмах, хирургических вмешательствах, в результате случайного заноса. Попадая в кровь, энтерококки могут колонизировать патологически измененные клапаны сердца, вызывая вялотекущий эндокардит (см. выше). К счастью, они обладают слабой вирулентностью, и энтерококковая бактериемия, которая не является редкостью, обычно не переходит в патологию.
С середины 90-х гг. получили распространение штаммы энтерококка, устойчивые к ванкомицину. Это явилось неприятным известием, во-первых, из-за сложностей лечения энтерококковых инфекций, а, во-вторых, из-за перспективы борьбы с антибиотикоустойчивыми стафилококками, для которых ванкомицин является препаратом выбора. Возникло опасение передачи транспозонов резистентности от энтерококков стафилококкам, тем более что в опытах in vitro это удалось сделать. Однако механизм природной устойчивости S. aureus к ванкомицину оказался иным, чем у энтерококка (см. «Стафилококки»). В основе энтерококкового варианта лежит изменение конечной структуры боковой пептидной цепи (d-Ala-d-Ala на d-Ala-d-лактат), что делает синтез пептидогликана (сшивание гликановых цепей) не чувствительным к ванкомицину. Тем не менее устойчивость к ванкомицину остается тревожной информацией, с которой пытаются бороться, опираясь, в частности, на эпидемиологию нозокомиальной энтерококковой инфекции.
Грамположительные анаэробные кокки
Это небольшая группа грамположительных бактерий, которые морфологически напоминают стрептококки (пептострептококки, около 10 видов) или стафилококки (пептококки, единственный вид), но не имеют с ними ничего общего. Они являются облигатными анаэробами, получившими свое название из-за способности ферментировать пептон и аминокислоты до уксусной кислоты (термины вряд ли можно считать удачными, так как пептон охотно утилизируют многие гетеротрофы). Присутствуют в микрофлоре здоровых людей (кишечник, влагалище, миндалины, десневые карманы), выделяются (обычно в смешанной культуре) из очагов гнойной инфекции брюшной полости (аппендицит, перитонит), ротовой полости (периодонтит, тонзиллит), генитальных органов, из раневых абсцессов. Внимание к пептострептококкам и пептококкам отражает возросший интерес к пиогенным осложнениям, связанным с неспорообразующими анаэробными бактериями, которые входят в состав нормальной микрофлоры человека. Здесь доминируют бактероиды, патогенетическая роль анаэробных кокков менее очевидна (см. «Неклостридиальная анаэробная инфекция»).
Пневмококк
- Микробиология и экология.
- Капсула и другие факторы болезнетворности.
- Пневмонии: проблема эндогенных и экзогенных инфекций.
- Внелегочные пиогенные инвазии: средний отит и менингит.
- Иммунологические подходы к профилактике пневмококковых инфекций.
- Антибиотикотерапия и резистентность пневмококка.
Правомерность включения пневмококка (Str. pneumoniae) в род Streptococcus разделяют не все микробиологи. Он существенно отличается от других стрептококков по ряду фенотипических и генотипических признаков, в том числе таких, которые определяют его взаимоотношения с человеком. Не случайно таксономическое положение пневмококка неоднократно менялось, и некоторые авторы не без оснований настаивают на его выделении хотя бы в самостоятельный род семейства Stpreptococcaceae.
В своем большинстве клетки пневмококка располагаются парами (отсюда историческое название — диплококк Френкеля—Вексельбаума, по авторам, выделившим его в 1886 г. от больных крупозной пневмонией) и защищены толстой полисахаридной капсулой, которая окружает оба кокка или образует чехол вокруг коротких цепочек (рис. 1). В классических описаниях форму пневмококковых клеток сравнивают с контурами пламени свечи или ланцета (ланцетовидный диплококк), хотя чаще (особенно в культурах) они выглядят как овальные кокки.

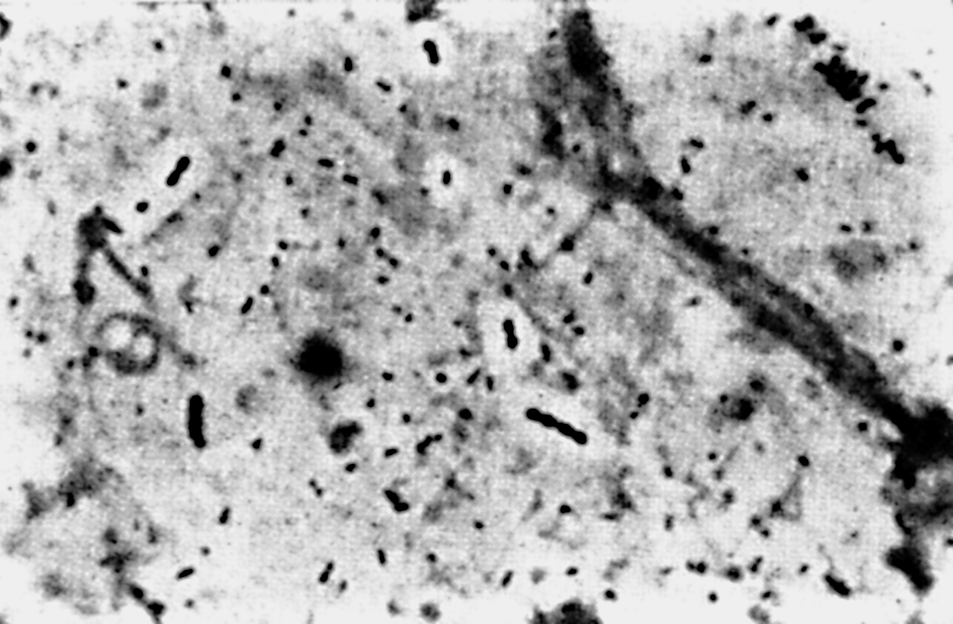
По культуральным признакам пневмококк похож на зеленящие стрептококки, но его нетрудно отличить по ряду других свойств, в частности по чувствительности к оптохину или по быстрому лизису поверхностноактивными веществами (например, желчными кислотами или просто желчью). Это связано с активацией аутолиза бактерий, а не с прямым деструктивным эффектом данных препаратов. Удивительная склонность пневмококка к аутолизу (она имеет значение для размножения бактерий) определяется ферментом LytA, разрушающим, как амидаза, бактериальный пептидогликан. Это может иметь патогенетическое значение, так как при лизисе пневмококков высвобождается холестеринсвязывающий белок, пневмолизин, обладающий способностью к повреждению альвеолярного эпителия и сосудистого эндотелия. Это служит причиной кровоизлияний (пурпура) и обильной экссудации в альвеолярное пространство при пневмококковой пневмонии. Из других эффектов пневмолизина отметим цилиостатическое действие (подавление активности реснитчатого эпителия), связывание с Fc-фрагментом антител (ведет к активации классического каскада комплемента), а также способность стимулировать макрофаги к выделению флогогенных цитокинов, прежде всего ФНО-a. Впрочем, значение пневмолизина в болезнетворности оспаривается тем, что капсульный серотип 3, один из самых вирулентных среди пневмококков, продуцирует низкий уровень данного белка.
Выделение пневмококка из носоглотки не редкость. Он периодически присутствует в верхних отделах респираторного тракта большинства здоровых людей. Первым, кто наблюдал пневмококк, был Л. Пастер. Он обнаружил его в слюне больного бешенством, заподозрив в причастности к заболеванию. Но от этого пришлось отказаться, так как идентичные бактерии определялись и у здоровых людей.
Пневмококк находится в абсолютной зависимости от своего хозяина и быстро погибает вне организма. Он весьма требователен к питательным средам (особенно при первичных посевах) и условиям культивирования. Поэтому самым надежным способом его выделения является внутрибрюшинное заражение белых мышей мокротой больного. Уже через несколько часов в брюшной полости появляется множество пневмококков, а через 24—48 ч животные погибают от сепсиса. Бактерии легко выделить из крови, селезенки и других органов. Таким же способом удается повысить вирулентность пневмококка, в частности усилить способность к капсулообразованию.
Симбиоз с пневмококком дается ценой серьезного напряжения иммунитетных ресурсов (см. «Стрептококки», рис. 5). Более того, существует до сих пор не опровергнутая (но и не доказанная) гипотеза о том, что бурное течение крупозной пневмонии, когда молниеносно захватывается доля и даже целое легкое, базируется на аллергии к пневмококку, т.е. на отягощенном иммунологическом анамнезе.
Пневмококки дифференцируют по антигенным особенностям капсульных полисахаридов (К-антигены). Известно более 80 К-сероваров (К-серотипов), идентификация которых представляет не только академический интерес, но и имеет эпидемиологическое значение, дополняя патогенетическую характеристику штаммов. Это связано с тем, что не все серовары играют одинаковую роль в патологии человека, а сопоставление сероваров пневмококковой аутофлоры зева с сероварами, изолированными из очагов поражения, позволяет отличить последствия активации эндогенных штаммов от экзогенного заражения.
Уникальными свойствами обладает так называемый С-полисахарид клеточной стенки пневмококка (он не имеет ничего общего с С-групповым полисахаридом стрептококков). Это тейхоевая кислота, которая благодаря своему фосфорил-холиновому фрагменту соединяется с одним из острофазных белков сыворотки крови, получившим по этой реакции название С-реактивного белка (СРБ). Реакция легко учитывается по образованию преципитата и используется в оценке активности воспалительных процессов. Сходным эффектом обладает F-антиген — липотейхоевая кислота пневмококковой клеточной стенки.
Пневмококк располагает рядом других поверхностных компонентов, которые обсуждаются прежде всего в связи с проблемами бактериальной адгезии и возможностью использования в качестве вакцинных препаратов. Они были обнаружены в опытах с моноклональными антителами и имеют слишком сложные названия, вынуждая пользоваться акронимами: PspA (pneumococcal surface protein A), РspС (pneumococcal surface protein C), PsaA (pneumococcal surface adhesin A), SpsA (S. pneumoniae surface protein A), CbpA (choline binding protein A). Некоторые из этих белков (PspA и PspC) обладают множеством антигенных вариантов, которые отличают штаммы даже в пределах одного капсульного серотипа, накладывая отпечаток на взаимоотношения с хозяином. Вариабельность становится важным фактором для понимания неоднозначности инфекций, вызываемых идентичными сероварами пневмококка. Вместе с тем, кроме дискретных эпитопов, пневмококковые белки имеют много общего, позволяя надеяться на создание унифицированных вакцин. Опыты на животных говорят о такой возможности.
Патология
Пневмонии. Специфической пневмококковой инфекцией является лобарная пневмония. Она больше известна как крупозная пневмония, так как альвеолярный экссудат богат фибрином, т.е. соответствует экссудату при крупозном воспалении. Болеют взрослые, реже — дети, у новорожденных заболевание не встречается. В 95% случаев причиной служит пневмококк. Процесс начинается остро и с поразительной скоростью охватывает значительную часть легкого. Иногда одновременно или последовательно поражаются несколько долей и даже оба легких. Экссудат с примесью эритроцитов заполняет альвеолы, вытесняя воздух и сокращая дыхательную поверхность легких (рис. 2).

Повышенное содержание фибрина делает экссудат и соответственно мокроту вязкими, затрудняя отхаркивание. Через несколько дней экссудат приобретает фибринозно-гнойный характер, альвеолы (благодаря действию хемокинов) заполняются нейтрофилами, которые вместе с неспецифическими (комплемент, СРБ) и специфическими (антитела) опсонинами обеспечивают уничтожение возбудителя и рассасывание фибринозно-геморрагического экссудата. Этому моменту соответствует кризис заболевания — падение температуры и начало клинического выздоровления. Очищение альвеол завершают макрофаги. Весь процесс занимает 2—3 нед. И без того тяжелое заболевание может осложниться септической бактериемией с развитием гнойного менингита, перикардита, отита, эндокардита, артритов, тромбофлебитов и пр. Из местных осложнений возможны нагноение плевры, абсцесс легкого. Однако все это — больше классическая дидактика, нежели клиническая реальность. Современная антимикробная терапия обрывает естественное течение крупозной пневмонии и практически исключает вероятность местных и общих септических осложнений. Сегодня типичная крупозная пневмония встречается редко и по распространенности значительно уступает бронхопневмонии.
Удивительно, но несмотря на тяжелое течение, больные крупозной пневмонией неконтагиозны и традиционно госпитализируются в общие палаты терапевтических стационаров. В сочетании с широкораспространенным носительством Str. pneumoniae это побуждало думать об эндогенной инфекции, связанной с активацией «нормальных» пневмококков респираторного тракта. Однако было установлено, что серотипы штаммов, изолированных из очагов поражения и из зева того же больного, часто не совпадают. Поэтому, не отрицая патогенетической значимости аутоштаммов, приходится признать, что во многих (если не в большинстве) случаях пневмококковые заболевания возникают в результате экзогенного заражения от носителей вирулентных разновидностей пневмококка. Вместе с тем взаимоотношения с пневмококком высокоиндивидуальны. Для большинства людей столкновение даже с избранными (с точки зрения вирулентности) штаммами остается без последствий; симптомы заболевания возникают в единичных случаях. С этой точки зрения, Str. pneumoniae ближе к условно-патогенным бактериям, хотя и способен вызывать инфекции, которые принято считать первичными.
Природа этого противоречия до сих пор окончательно не раскрыта, хотя можно выделить ряд условий, предрасполагающих к развитию пневмонии. Вероятность клинического проявления респираторной инфекции обратно пропорциональна состоянию колонизационной резистентности бронхолегочного тракта. Она складывается из суммы факторов и механизмов, важнейшими из которых являются мукоцилиарный транспорт, обеспечивающий вымывание с током слизи чужеродных частиц; макрофаги, встроенные в стенку альвеол и бронхов; компоненты слизи, блокирующие адгезию бактерий и экранирующие адгезины эпителиоцитов; бактерицидные и бактериостатические факторы слизи (лизоцим, лактоферин, миелопероксидаза и др.). В норме эти механизмы обеспечивают стерильность терминальных бронхов и альвеол, несмотря на постоянную ингаляцию зараженного воздуха. Только поступление большого количества зараженного материала (например, при слабом кашлевом рефлексе у лиц в состоянии глубокого опьянения) или ослабление клиринговых ресурсов местного иммунитета (повреждение эпителия респираторного тракта вирусами, курением, профессиональными вредностями, например угольной пылью) могут привести к стабилизации инфекта и развитию пневмонии.
Бронхопневмония — обычно вторичная инфекция, которая наслаивается на вирусные респираторные инфекции, коклюш, хронический бронхит, сердечно-сосудистые заболевания (отек легких), тяжелые оперативные вмешательства, переохлаждение. В качестве негоспитальной («бытовой») инфекции чаще встречается у лиц пожилого и старческого возраста (отсюда ироничное название — друг стариков). В отличие от крупозной пневмонии, когда экссудат быстро распространяется через поры в межальвеолярных перегородках на всю долю легкого, при бронхопневмонии очаги воспаления существуют раздельно (их размер составляет около 1 см). Бронхи с прилегающими альвеолами заполняются фибринозным экссудатом, содержащим множество нейтрофилов. Пневмококк — обычная причина бронхопневмонии, хотя этиологический профиль здесь далеко не столь однороден, как при крупозной пневмонии. Нередко приходится сталкиваться со стафилококками, стрептококками, палочкой инфлюэнцы, клебсиеллами. Инфекция может быть смешанной.
Внелегочные инвазии. Вексельбаум и Френкель, утвердившие патогенетическую значимость пневмококка, полагали, что пневмония — единственная пневмококковая инфекция. К сожалению, они ошибались. Воспаление легких — эталонная, но не самая распространенная инфекция, вызываемая Str. pneumoniae. Два пиогенных процесса доминируют в послужном списке этих бактерий: средний отит и менингит (рис. 3). Они чаще встречаются у детей младшего возраста, хотя и для взрослых (особенно менингит) представляют реальную опасность. Это может быть связано с типичной для грамположительных бактерий устойчивостью к литическому эффекту комплемента, который слабо или совсем не проявляется даже в присутствии антител, а потому не защищает от внутрисосудистой инвазии. Гнойным средним отитом болеют около 20% детей, а, по некоторым данным, более 70% детей до трех лет хотя бы однажды имели абортивные признаки этого заболевания (см. таблицу). Инфекция проникает в среднее ухо из носоглотки по евстахиевой трубе и имеет опасную тенденцию к распространению на менингеальную оболочку. Но менингит возникает и как самостоятельное заболевание на фоне эпизодической пневмококковой бактериемии. По данным мировой статистики, средний отит и менингит (в периоды эпидемического благополучия по менингококковой инфекции) определяют, наряду с Haemophilus influenzae, лидерство пневмококка в современной клинике пиогенных бактериальных инфекций у детей (более подробно см. «Палочка инфлюэнцы»).

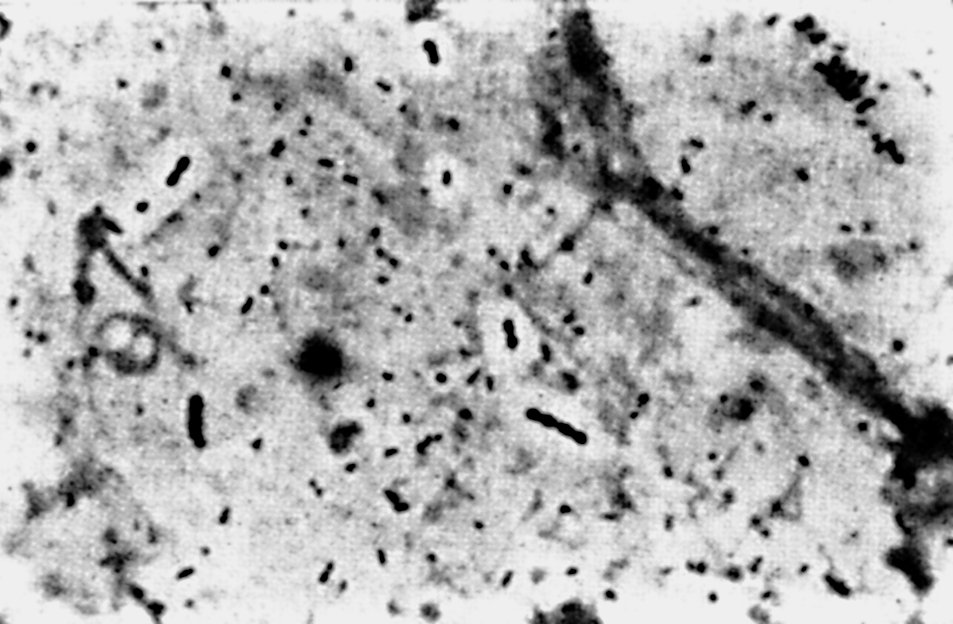
Внутрисосудистая инвазия, которая является отнюдь не редким событием в естественных взаимоотношениях с пневмококком, может стать причиной и ряда других пиогенных осложнений — артрита, эндокардита, перитонита (чаще у девочек). Вероятность каждого из них возрастает, если в организме уже имеется очаг пневмококковой инфекции (пневмония, отит, менингит).
Бактерии, выделяемые при остром пиогенном среднем отите у детей*
| Бактерии | Положительный результат, % |
| Str. pneumoniae H. influenzae Str. рyogenes (группа А) S. aureus Moraxella catarrhalis Cмешанная флора Отсутствие или выделение непатогенных бактерий |
39 20 |
*Mandell, Douglas and Bennetts. Principles and Practice of Infectious Diseases. 4th ed. Churcill & Livingstone, 1995.
Факторы вирулентности
Природа болезнетворности пневмококка во многом остается загадкой. Понятна лишь причина его высокой инвазивности. Она связана с капсулой, которая позволяет противостоять фагоцитам и комплементу до тех пор, пока бактерии не получат подкрепления в виде специфических опсонинов, т.е. антител. Это доказывают результаты экспериментов на животных (авирулентность бескапсульных штаммов, протективность противокапсульных антител), а также наблюдения за клинической эффективностью специфической серотерапии при крупозной пневмонии. Применение сывороток животных с высоким содержанием антикапсульных антител способствовало выздоровлению и снижало вероятность осложнений.
Кроме факторов, о которых говорилось выше, у пневмококка обнаружены гиалуронидаза и нейраминидаза — ферменты, разрушающие основное вещество соединительной ткани. Они способствуют расширению зоны инфекции, но их значение гораздо ниже инвазивности, детерминируемой капсулой. Пневмококки, как и ряд других бактерий, паразитирующих на слизистых оболочках, располагают специфической IgA-протеазой. Она способна разрушать антитела, подавляющие адгезию бактерий, хотя реальное значение подобных ферментов в развитии инфекции остается под вопросом.
Не ясно и то, каким образом пневмококки закрепляются в инфицированных тканях. Известно лишь, что в адгезии на эпителиоцитах верхних дыхательных путей (в носоглотке) и на пневмоцитах/эндотелиоцитах задействованы разные бактериальные структуры, имеющие сродство к дискретным гликоконъюгатам — гликолипидам и гликопротеинам. В первом случае в состав клеточных рецепторов входят дисахариды семейства неолактозы, во втором — углеводные радикалы более разнообразной структуры, содержащие маннозу, производные галактозы и др. Замечено, что женское молоко, содержащее неолактозные гликоконъюгаты, ослабляет закрепление пневмококка на эпителии носоглотки. Это позволяет думать о профилактическом эффекте грудного вскармливания в отношении пневмококковых инфекций, прежде всего среднего отита.
Мало что можно сказать и о природе токсического компонента инфекции. У пневмококка обнаружены гемолизины и лейкоцидины, однако они не могут претендовать на роль специфического токсина пневмококков. Капсульный материал также не токсичен для животных и безвреден при введении в кожу человека.
Одним из существенных признаков пневмококкового поражения является крупозный характер воспаления. Это связано с повреждением эндотелия, после чего он начинает пропускать плазму и даже эритроциты (фибринозно-геморрагический экссудат). Выше говорилось, что поиски фактора, который способен повреждать стенку сосудов и вызывать кровоизлияния, привели к обнаружению пурпурогенных свойств у продуктов аутолиза пневмококка. Такой активностью обладают пневмолизин и производные пептидогликана, известные как пурпурогенин. Обсуждается роль перекиси водорода, которая в большом количестве продуцируется пневмококком и в отсутствие каталазы оказывает мембранолитический эффект на эндотелиальные клетки и альвеолоциты. Но в целом Str. pneumoniae относится к бактериям со слабой токсигенностью. С этой точки зрения, проявления тяжелой интоксикации при пневмококковых инфекциях следует считать вторичными: они отражают реакцию на внедрение возбудителя и самоотравление организма эндогенными продуктами, в частности цитокинами активированных макрофагов. Обсуждая патогенетически значимые факторы пневмококка, нельзя обойти и тот факт, что болезнетворность его капсульных сероваров неодинакова. Неодинаково и участие отдельных серотипов в развитии разных пневмококковых заболеваний, например пневмонии и отита. Большинство пневмоний связано с сероварами 1—4, 6—8, 14, 18 и 19. С известной определенностью можно говорить лишь о причине повышенной вирулентности серовара 3. Пневмококки этого типа наиболее активно продуцируют капсульное вещество с актикомплементарными свойствами, а потому надежнее других защищены от фагоцитоза. Неодинаковая вирулентность штаммов, по-видимому, не связана с особенностями их адгезивных реакций: способность Str. pneumoniae прилипать к эпителию не зависит от К-типовой принадлежности.
Иммунитет и вакцины
Перенесенная инфекция оставляет типоспецифический иммунитет, детерминируемый противокапсульными антителами-опсонинами. Иммунитет — непрочный, после крупозной пневмонии он держится 6—12 мес. Разработана вакцина на основе капсульных полисахаридов из 14 наиболее важных в патогенетическом отношении пневмококков. Однако ее эффективность и целесообразность применения не бесспорны. Пневмококковые полисахариды относятся к Т-независимым антигенам. Поэтому их иммуногенность и оставляемая ими иммунологическая память невелики, особенно у детей младшего возраста. Наряду с серологическим многообразием пневмококка это объясняет то, что чувствительность к пневмококку, причем не только к внутрилегочной, но и к внутрисосудистой (менингит!) инвазии, сохраняется на протяжении всей жизни.
Предпринимаются попытки усилить вакцинальный эффект полисахаридов путем их конъюгации с белковыми носителями, в частности с дифтерийным и столбнячным анатоксинами. В сферу внимания попали и поверхностные белки пневмококка. Некоторые из них выполняют функцию адгезинов, и антитела к ним в опытах на животных оказывают протективный эффект. Кроме того, наряду с антигенной вариабельностью они содержат консервативные эпитопы, что позволяет надеяться на унификацию иммуногенного начала.
Антибиотикотерапия
Подобно пиогенному стрептококку, пневмококк сохраняет чувствительность к антибиотикам, в том числе к препаратам пенициллина, которые многими продолжают рассматриваться как лучшее средство для лечения пневмококковых инфекций. Вместе с тем, начиная с 1960-х гг., появилось немало сообщений о выделении бета-лактамустойчивых штаммов и о вариантах, резистентных к другим антибиотикам (тетрациклины, макролиды, аминогликозиды, хлорамфеникол). Это грозить стать большой проблемой, побуждая задумываться о возрождении противопневмококковой серотерапии. В отличие от большинства стафилококков, механизм устойчивости пневмококка к пенициллину и другим бета-лактамам не связан с продукцией бета-лактамаз — ферментов, разрушающих антибиотики. Здесь речь идет об изменениях мембранных компонентов, взаимодействующих с пенициллином. Это так называемые пенициллинсвязывающие белки, которые фукционируют как ферменты, синтезирующие пневмококковый пептидогликан. Ситуация напоминает резистентность к ванкомицину у метициллинрезистентных золотистых стафилококков (см. «Стафилококки»). В этом случае генетическая информация, по-видимому, передается от оральных стрептококков, адаптированных к различным антибиотикам.
В 1986 г., когда отмечалось столетие со дня открытия пневмококка, летальность от пневмококковых инфекций превосходила число фатальных исходов при всех остальных бактериальных инфекциях, вместе взятых. Об этом печальном лидерстве не следует забывать и сегодня. Пневмококк остается одной из главных причин инфекционной патологии и смертности во всем мире. Этому способствует распространение клонов, устойчивых к антибиотикам.
Менингококк
Ни одна из инфекций не убивает так быстро.
W.W. Herrick
- Лидер среди лидеров.
- Серогруппы, серовары и клонотипы.
- От носительства до молниеносного сепсиса.
- Условия и факторы внутрисосудистой инвазии.
- Патогенез и диагностика менингококковой инфекции.
- Дихотомия местного и системного иммунитета.
- Проблемы вакцинопрофилактики и антибиотикотерапии.
К наиболее драматичным заболеваниям, безусловно, принадлежит гнойный цереброспинальный менингит — пиогенное воспаление оболочек головного и спинного мозга. В типичных случаях он протекает с симптомами глубокого поражения центральной нервной системы на фоне тяжелейшей интоксикации. До появления сульфаниламидов и, особенно, пенициллина смертность достигала 70%, хотя и сегодня гнойный менингит занимает одно из первых мест по числу летальных и инвалидизирующих осложнений, особенно среди детей младшего возраста. Каждый случай гнойного менингита — огромное испытание для больного, его близких и врачующего персонала. Отсюда понятно, каким тревожным событием являются вспышки, а тем более эпидемии этого заболевания.
Тяжелое, нередко молниеносное течение, неуверенность в выздоровлении, угроза непредвиденного заражения сеяли панику, доходившую до истерии. Эпидемии гнойного менингита (к счастью, они всегда носили ограниченный характер, не переходя в пандемии) неоднократно поражали человечество, и именно во время одной из них, в конце XIX в., был выделен возбудитель. Это сделал в 1887 г. австрийский патолог и военный хирург А. Вексельбаум, но потребовалось еще десятилетие, чтобы болезнетворность менингококка стала общепризнанным фактом. Современное название Neisseria meningitidis появилось много позже. Оно отражает принадлежность к роду Neisseria семейства Neisseriaceae (в честь немецкого микробиолога А. Нейссера), а видовой эпитет подчеркивает исключительное право менингококка называться возбудителем эпидемического цереброспинального менингита.
Мозговые оболочки поражаются различными бактериями, вирусами, реже — другими микробами. Наряду с менингококком (символом пиогенной инвазии центральной нервной системы) к возбудителям гнойного менингита относится еще два вида бактерий — Streptococcus pneumoniae и Haemophilus influenzae (см. соответствующие очерки). Их значимость, согласно разным данным, колеблется, но есть закономерности (прежде всего возрастные и эпидемиологические), которые характерны для каждого из представителей этой печально знаменитой тройки. Например, пневмококк чаще поражает взрослых, тогда как N. meningitidis и, особенно, H. influenzae больше нацелены на младший детский контингент. Пневмококковые и гемофильные менингиты иногда дают групповые вспышки, но они всегда эндемичны, не достигают масштаба эпидемий. Эпидемический менингит — привилегия менингококка, а если быть точнее, его так называемых эпидемических штаммов, которые относятся к определенным серологическим вариантам, или, точнее, клонам.


Подобно всем нейссериям, менингококки представляют собой мелкие уплощенные кокки (в виде кофейного зерна), собранные в большинстве попарно (рис. 1). Они грамотрицательны, неподвижны, спор не образуют. Имеют пили, хотя встречаются и беспилевые варианты. Образование пилей удается контролировать в фазовых вариантах, подбирая соответствующие условия культивирования (рис. 2). Пили отличаются высокой структурной и, следовательно, антигенной изменчивостью, которая происходит за счет внутрихромосомных рекомбинаций с «молчащими» последовательностями гена пилина PilS (от англ. silent — молчащий). Это обусловливает несостоятельность идеи создания на основе пилей антиколонизационной вакцины, способной заблокировать инфекционный процесс в его самом начале.

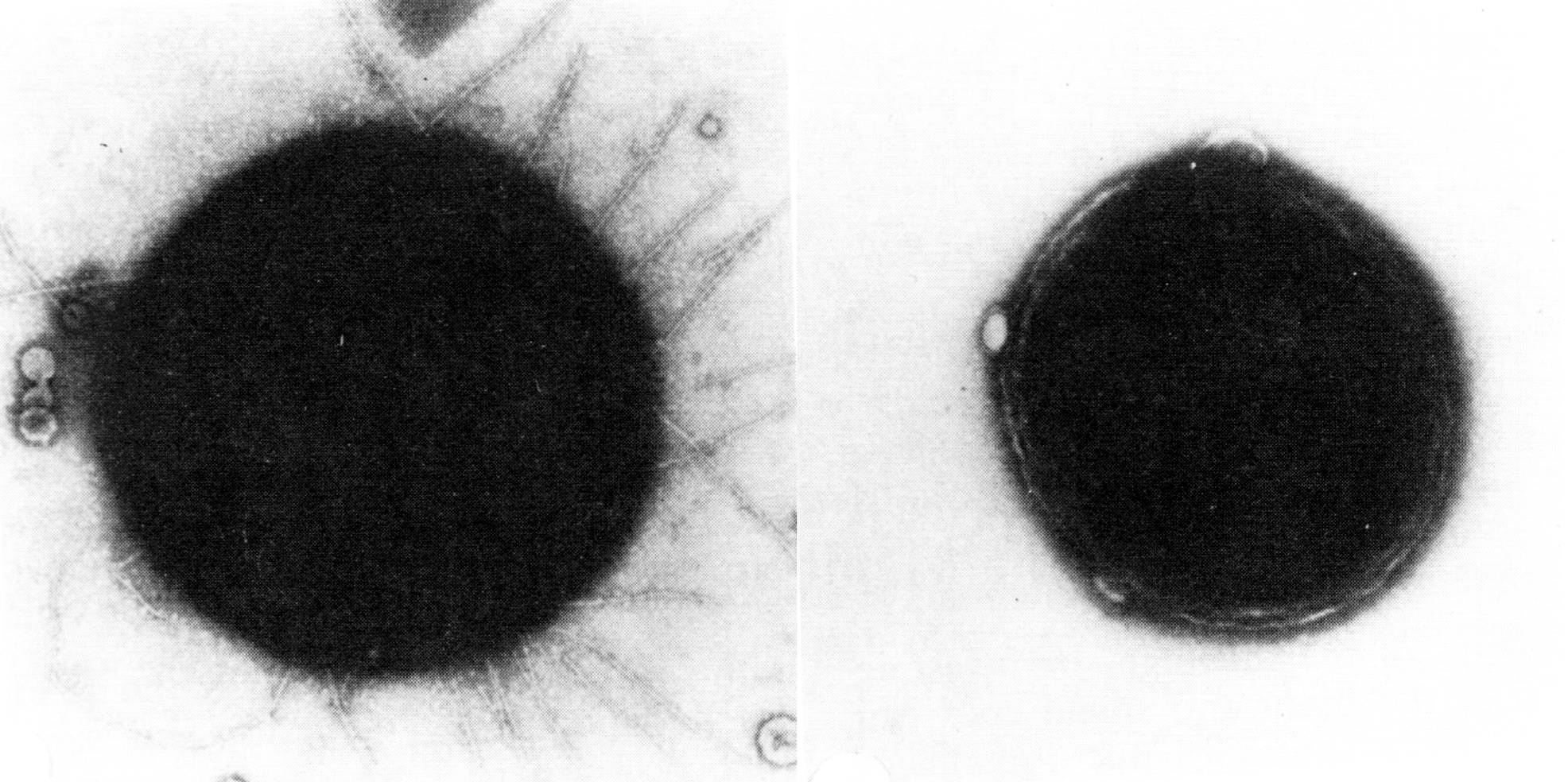
По строению клеточной стенки менингококк не отличается от других грамотрицательных бактерий. Патогенетически важным компонентом является наружная мембрана, которая как и у других грамотрицательных бактерий состоит из липополисахарида и поверхностных белков. Доминирующие белки наружной мембраны включают 5 классов: 1 — PorA; 2,3 — PorB; 4 — Rmp; 5 — Ора и Орс. Белки PorB (20 серотипов) и PorА (10 субтипов) встраиваются в мембрану эукариотической клетки, содействуя реорганизации актина и внутриэпителиальной инвазии. Ора и Орс отличаются высокой индивидуальной вариабельностью. Их антигенная пластичность связана с обратимыми мутациями в генах, что играет существенную роль в адаптивных реакциях менингококков, содействуя ускользанию от приобретенных в прошлом антител. Акронимы Ора и Орс возникли из английского слова opacity (мутность). Дело в том, что штаммы, у которых определяются подобные белки, формируют мутные колонии. Они бедны капсульным материалом и обычно выделяются из носоглотки носителей, т.е. в условиях, требующих селекции против сформировавшегося иммунитета.
В наружной мембране менингококка имеются минорные белки, конкурирующие за ионы железа — рецепторы трансферрина (Tbp A/B), лактоферрина (Lbp A/B) и гемоглобина (Hbm R, Hpu A/B). Кроме того, менингококки используют железо из сидерофоров индигенной флоры, а также извлекают его из гема и комплекса гемоглобина с гаптоглобином. Это важный механизм патогенности, так как железо необходимо для реализации окислительно-восстановительного потенциала, с которым связаны многие функции бактерий. Следует заметить, что аналогичные «железодобывающие» системы имеются у гонококка и других (непатогенных и условно-патогенных) нейссерий, обеспечивая их выживание и агрессивность в организме хозяина. Cигналом для усиления экспрессии рецепторов является увеличение железа во внешней среде. Значение данного механизма демонстрируют опыты по значительному (в 1000 раз) снижению для мышей вирулентности менингококков, выращенных на среде без источников железа.
Менингококковый липополисахарид отличается рядом структурных особенностей, усиливающих его биологическую (патогенетическую) активность. Его часто называют липоолигосахаридом, так как он построен лишь из базисной части — липида А и кор-углевода (см. «Болезнетворность бактерий»). В нем отсутствуют боковые углеводные радикалы, которые детерминируют эпитопное многообразие липополисахаридных антигенов (О-специфичность) и в известной мере экранируют биологически активный фрагмент молекулы, липид А. Такая структура типична для липополисахаридов R-форм. У большинства бактерий R-формы образуются в результате патологической изменчивости, тогда как для менингококка это видовая норма. R-липополисахариды близки (но не идентичны) в антигенном отношении. Этим отчасти объясняются антигенное родство всех менингококков и их антигенные связи с другими грамотрицательными бактериями — феномен, который вносит определенный вклад в патогенез менингококковых инфекций (см. ниже). Имеет значение и то, что липополисахаридный эндотоксин менингококка сиалирован. Присутствие сиаловой (нейраминовой) кислоты обеспечивает резистентность к альтернативному пути активации комплемента, что является важным фактором устойчивости бактерий в сосудистом русле. Заметим также, что при росте менингококки сбрасывают свои мембранные компоненты в виде пузырьков (англ. blebs). Это содействует выделению значительных количеств эндотоксина, который входит в состав таких пузырьков. По особенностям липополисахарида дифференцируют 13 иммунотипов менингококка. В антигенной формуле штамма их обозначают буквой L и соответствующим номером.
Снаружи менингококки покрыты полисахаридной капсулой. На основании ее антигенных особенностей дифференцируют серологическиe группы: A, B, C, D, E (29E), I, K, X,Y, W(W135), Z. Более 90% инфекций связано с группами А, В и С. Штаммы группы А ответственны за большинство крупных эпидемических вспышек, группы В и (в меньшей степени) С превалируют в межэпидемические периоды, вызывая спорадические заболевания и ограниченные вспышки в небольших коллективах. Впрочем, иногда ситуация меняется, и в эпидемический процесс включаются другие менингококки.
По антигенным особенностям белков наружной мембраны большинство серогрупп можно дополнительно разделить на серотипы (1—20, PorB) и субтипы (1—10, PorA). Часть из них ограничена определенными группами, другие — представлены в разных серогруппах. Примером такого серотипирования является В:4:Р1 — штамм серогруппы B, серотипа (PorB) 4, субтипа (PorA) P1. Для описания антигенных свойств липополисахарида добавляется буква L c конкретным (1—13) номером.
Некоторые из типовых (субтиповых) антигенов предложено использовать в качестве маркеров эпидемичности, хотя причины, лежащие в основе неодинаковой вирулентности менингококковых штаммов, не известны. Для эндемичных и спорадических инфекций такой зависимости не обнаружено. Серотипирование помогает в развитии вакцинальной стратегии, но не дает возможности решать эпидемиологические вопросы.
Главную роль в изучении закономерностей распространения менингококковой инфекции играет клонирование штаммов на основе современных методов генетического анализа. Это позволяет судить не только о вариабельности единичных генов, но и давать заключение о множественных изменениях хромосомных «домашних» (англ. housekeeping) генов, констатируя их внедрение в менингококковый процесс. N. meningitidis относится к представителям поликлональных бактерий. Из многих генотипов серогруппы А лишь отдельные клоны вызывают крупные эпидемии. Это характерно и для других серогрупп менингококка. Группы В и С имеют сходные с менингококком А закономерности распространения и внутригрупповую неоднородность клонов по вирулентности.
Менингококк принадлежит к числу прихотливых и трудных для культивирования бактерий. Его требуется выращивать на средах с добавлением нативных биологических субстратов (кровь, сыворотка, асцитическая жидкость) либо на сложных синтетических средах. Множественная ауксотрофность менингококка (т.е. зависимость от ростовых факторов) позволяет легко дифференцировать его от непатогенных нейссерий, постоянно обитающих в носоглотке. Последние, в отличие от N. meningitidis, растут на простых питательных средах при комнатной температуре; менингококк же не размножается при температуре ниже 25оС. Подобно всем нейссериям, менингококк относится к аэробам, хотя и размножается лучше в атмосфере 3—10% CО2. Как и другие аэробы, он продуцирует оксидазу, по которой его колонии легко распознать при первичных посевах из клинического материала (рис. 3).



Рис. 3. Идентификация колоний менингококка по оксидазному тесту. Смешанная культура на кровяном агаре: а — колония менингококка и контаминирующих бактерий перед внесением 1% раствора триметил-р-фенилендиамина; б — та же культура после добавления реагента; колонии N.meningitidis выглядят темными, особенно по краям
Менингококки склонны к аутолизу, что, по-видимому, имеет патогенетическое значение. Это проявляется и in vitro. Поэтому культуры необходимо часто пересевать (субкультивировать), иначе они гибнут. Не удивительно, что менингококк весьма неустойчив во внешней среде. Он быстро погибает в стандартных растворах дезинфектантов, при высыхании, чувствителен к высоким и низким температурам и, в отличие от большинства бактерий, быстро теряет жизнеспособность в обычном холодильнике. Перечисленные признаки (прихотливость в питании, неустойчивость во внешней среде, которая, кстати, исключает необходимость дезинфекционных мероприятий в очагах инфекции) сближают N. meningitidis с гонококком (см. «Гонококк»).
В естественных условиях менингококк выделяется только от человека. Самой распространенной формой менингококковой инфекции является носительство бактерий в носоглотке. По усредненным данным, около 10% здоровых людей инфицированы менингококком. Обычно это кратковременное явление, не имеющее последствий для носителя и его окружения, тем более что 9/10 штаммов принадлежат к невирулентным клонам. Но в 5% случаев носительство становится злостным (длительное, иногда многолетнее и обильное инфицирование). Это серьезно осложняет ситуацию, тем более что такое носительство часто связано с эпидемическими штаммами групп А, В и С, доминирующих при инвазивных менингококковых поражениях. Носительство может осложниться очаговым воспалительным процессом — назофарингитом, локальной формой клинически значимой менингококковой инфекции. Как правило, это относительно безобидный процесс, но бактерии иногда проникают в кровь, что существенно осложняет прогноз. Кроме того, назофарингит сопровождается усиленным размножением бактерий, повышая вероятность контаминации окружающих.
Менингококк способен не только выживать, но и размножаться в кровяном русле (менингококцемия) и, оседая в различных тканях, инициировать генерализованный септический процесс. Особенно часто поражаются оболочки (мягкая, арахноидальная) головного и спинного мозга с развитием гнойного менингита. В процесс вовлекаются и другие органы (эндокард, надпочечники, кожа, суставы), чем определяется общая картина менингококкового сепсиса, т.е. генерализованной менингококковой инфекции. Парадоксально, но бессимптомные носители и больные менингококковым назофарингитом сами редко страдают столь серьезно, так как защищены от внутрисосудистой инвазии сывороточными антителами. Чаще они становятся рассадником воздушно-капельной инфекции, чреватой серьезными последствиями для окружающих. К счастью, и это случается редко: лишь в небольшом проценте случаев (по разным данным, 1:200—1:2000) очаговый менингококковый процесс, возникший в результате экзогенного инфицирования, эволюционирует в гнойный менингит и (еще реже) в молниеносные варианты септицемии. Есть данные, что в межэпидемический период на одного больного с генерализованной менингококковой инфекцией приходится (в год) до 50000 носителей. Возможно, что это тоже связано с естественным клонированием, т.е. с пластичностью штаммов, которая помогает им выживать в виде гетерогенных популяций в условиях коллективного иммунитета. Внутри каждой серогруппы в носоглотке носителей периодически возникают клоны, склонные к извазии, которые, проникая в чувствительный организм, способны вызывать менингококцемию.
Менингококк наиболее опасен для детей младшего возраста, особенно до года; с возрастом заболеваемость падает. Распределение носительства имеет противоположную направленность, увеличиваясь с возрастом. Угроза повышается при скученности населения, т.е. при тесных и постоянных контактах, допускающих интенсивный обмен микрофлорой. Высокий процент носителей (30% и более) рассматривается как тревожный сигнал, повышающий (особенно при большой скученности) шансы менингококка на столкновение с чувствительным организмом, т. е. с теми, кто впервые попал в данную популяцию и лишен иммунитета против циркулирующего в ней штамма. Этим объясняются внутриказарменные вспышки менингита среди солдат (прежде всего новобранцев), относящиеся в основном к периоду мировых войн. Впрочем, не все согласны с подобной логикой, так как корреляция между уровнем назофарингеального носительства менингококков и заболеваемостью менингитом отсутствует. Приспосабливаясь, менингококк оставляет невирулентные клоны, присутствие которых не говорит об опасности эндемичной (а тем более эпидемичной) инфекции.
Итак, патогенез менингококковой инфекции можно анализировать по трем главным направлениям: 1) стабилизация инфекции в носоглотке (бессимптомное носительство, назофарингит), 2) внутрисосудистая инвазия, септическая интоксикация и пиогенные метастазы во внутренние органы (менингококцемия), 3) инфекция мозговых оболочек (гнойный менингит).
Колонизация носоглотки инициируется связыванием менингококковых пилей с эпителиоцитами назофарингеального отдела респираторного тракта. Рецептором для пилина является белок CD46, защищающий многие клетки от действия собственного мембраноатакующего комплекса комплемента. Менингококк довольно избирательно взаимодействует с эпителиоцитами именно этой зоны слизистых оболочек. Прикрепление к буккальному эпителию, эпителиальным клеткам носовых ходов, уретры и мочевого пузыря происходит гораздо хуже. Контакт с пилином готовит условия для инвазии менингококка, в которой главную роль играют наружные мембранные белки Ора и Орс. Ора связывается с белковым рецептором семейства CD66, Орс — с поверхностными гепарансульфатпротеогликанами. Благодаря сокращению пилей, происходит физическое и функциональное сближение клетки и менингококка, которое заканчивается пенетрацией бактерий сквозь эпителий. Эффектором инвазии считается Орс, так как именно он способствует проникновению менингококков в эпителиоциты. Интересно, что этому мешает присутствие капсулы и сиалированного липоолигосахарида, которые обязательны для выживания менингококков в кровяном русле. Пилезависимый контакт с эпителиальными клетками индуцирует обратимую утрату этих атрибутов, сохраняя способность к реверсии при благополучном завершении трансцитоза. Вообще, в процессе носительства и инвазии экспрессия пилей, пятого класса наружных мембранных белков, капсулы и липополисахарида (липоолигосахарида) серьезно меняется в силу фазных (включить/выключить) и антигенных вариаций. Это те механизмы, которые могут быть использованы бактериями для ускользания от растущего иммунитета хозяина.
Инвазия в слизистую оболочку вызывает воспалительную реакцию — назофарингит, который обычно протекает как «мягкая» ангина. Впрочем, местный процесс напрямую не связан с вероятностью проникновения бактерий в кровь: внутрисосудистая инвазия наблюдается и без симптомов назофарингита. Допускают, что инвазивными свойствами (инвазивным фенотипом) обладают единичные клоны, возникающие в результате антигенных вариаций пилей и поверхностных белков, которые повышают тканевой тропизм и проникновение бактерий в эпителиоциты. Кроме активной инвазии, возможно и пассивное внедрение менингококков в глубжележащие ткани. Замечено, например, что менингококковым менингитом чаще болеют поздней осенью и ранней весной, когда число простудных заболеваний особенно велико. Полагают, что катаральные явления в носоглотке, вызванные другими микробами, способствуют инвазии менингококка, повышая риск системных осложнений. Это справедливо и для двух других наиболее распространенных возбудителей гнойного менингита — Haemophilus influenzae и Streptococcus pneumoniae.
Для стабилизации внутрисосудистого инфекта менингококки должны прежде всего выжить, преодолев бактерицидный барьер крови. Подобно другим грамотрицательным бактериям, менингококк чувствителен к литическому действию комплемента. В отсутствие протективных (антикапсульных) антител комплемент — единственное оружие против менингококцемии. Его активация в этом случае происходит через систему маннозосвязывающего белка или по альтернативному (антителонезависимому) пути, дефекты которых предрасполагают к менингококковым осложнениям. Впрочем, и сам менингококк неоднороден по чувствительности к комплементу. Например, капсула группы В состоит из сиаловой кислоты и поэтому слабо активирует альтернативный каскад комплемента. То же самое можно сказать о липополисахариде (липоолигосахариде) менингококков, который часто включает остатки сиаловой кислоты хозяина. Не случайно инфицирование такими штаммами людей, лишенных антител, повышает вероятность тяжелых осложнений.
Циркулирующие антитела против капсульных антигенов служат надежной гарантией против генерализации инфекционного процесса, но их протективность целиком зависит от комплемента. После связывания с бактериями они запускают его классический каскад, что обеспечивает максимальный бактерицидный эффект. Дефект по терминальным (С8—С9) факторам комплемента предрасполагает к менингококцемии и рассматривается как фактор риска для этой патологии. Как уже говорилось, среди белков наружной мембраны менингококка имеется протеин Rmp, антитела к которому ведут себя своеобразно. Взаимодействуя с антигеном (Rmp), они блокируют реакцию с антителами против липополисахарида и Por, защищая бактерии от их бактерицидного (активация комплемента) действия.
Присутствие противоменингококковых антител в крови у взрослых — обычное явление. Стимулом к их образованию служит скрытая иммунизация при здоровом (или почти здоровом) транзиторном носительстве менингококка, через которое к зрелому возрасту проходит большинство людей. Нехваткой антител можно объяснить повышенную чувствительность к менингококку детей первых лет жизни. При этом следует учитывать, что, хотя новорожденные и получают антитела от матери, но IgM-антитела (наиболее мощные активаторы комплемента) не проходят через плаценту. Парадокс, но бактерицидность комплемента может блокироваться самими антителами. Это известно для IgA-антител: не обладая способностью активировать комплемент, они защищают бактерии от комплементзависимой бактерицидности IgM- и IgG-антител. Индуктором образования антименингококковых блокирующих IgA-антител служит не только сам менингококк, но и антигены нормальной микрофлоры (прежде всего энтеробактерий), располагающие общими эпитопами с его поверхностными антигенами.
Еще одним парадоксом является разобщение системного и местного иммунитета. Циркулирующие противокапсульные антитела, эффективно защищающие от менингококцемии, не препятствуют развитию местной инфекции. Поэтому вероятность носительства менингококка и его локальных осложнений (назофарингит) не коррелируют с уровнем сывороточных антител. Кстати, вакцинация не снижает число носителей менингококка и не предупреждает заражения ими невакцинированных людей.
Из сказанного следует, что в крови менингококк ожидают серьезные испытания, пройти через которые ему удается далеко не всегда. Если это все-таки происходит, менингококк размножается, создавая угрозу системных септических осложнений.
Менингококки не располагают какими-либо особыми токсинами, и главным (возможно, единственным) фактором патогенности на этапе менингококцемии является липополисахаридный (точнее, липоолигосахаридный) эндотоксин. Его токсичность усиливается благодаря отсутствию О-специфических углеводных радикалов, экранирующих биологически активный фрагмент (липид А). В таком голом виде липополисахариды легче связываются с рецепторами клеток-эффекторов (прежде всего макрофагами) и гуморальными факторами, опосредующими это взаимодействие.
Но главное в том, что, преодолев бактерицидность крови, менингококк размножается и, накапливаясь в огромных количествах, высвобождает большую массу эндотоксина в виде мембранных пузырьков. Этому способствуют склонность менингококка к аутолизу и комплементзависимый бактериолиз. Гиперэндотоксинемия ведет к острейшему синдрому внутрисосудистой активации крови, характерному для септических состояний (см. «Сепсис»). Обычным спутником менингококцемии является геморрагическая сыпь (рис. 4), реже возникают молниеносный коллапс и смерть из-за тромботического некроза надпочечников (синдром Уотерхауза—Фридрихсена). В пораженных участках (например, в очагах кожной сыпи) обнаруживаются менингококки, которые благодаря кровоизлияниям из тромбированных сосудов получают доступ в ткани.

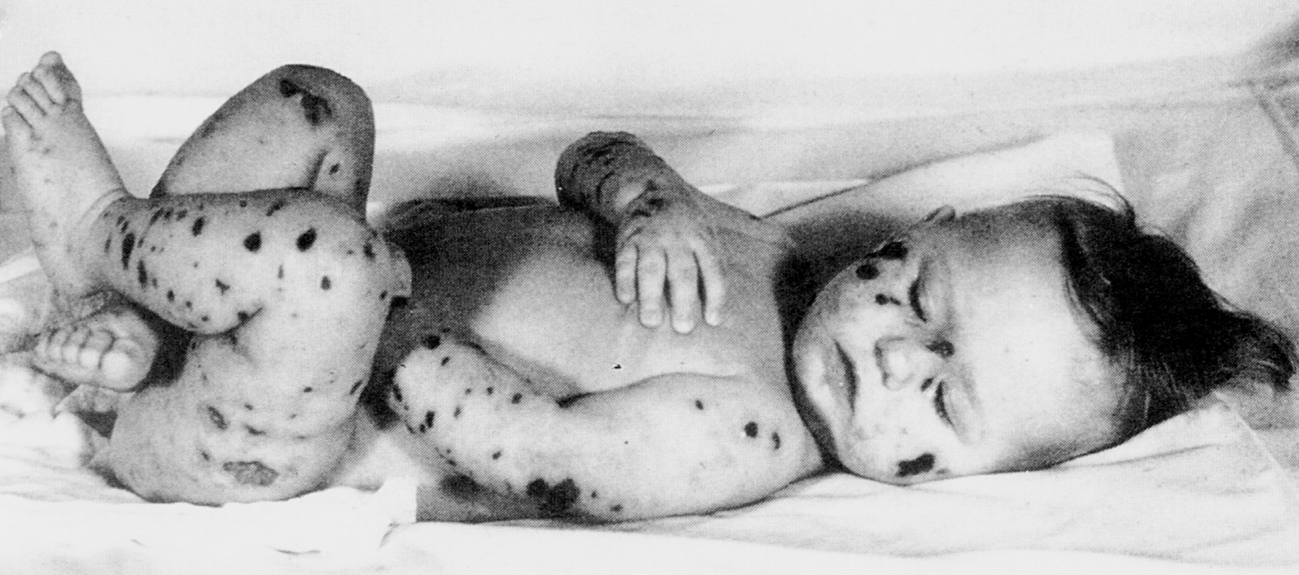
Поражение мозга по сути отражает сходный тромбогеморрагический процесс в сосудах мозговых оболочек. В цереброспинальной жидкости бактерии находят отличные условия для размножения. Здесь достаточное количество питательных веществ, но практически нет опсонинов (комплемента, антител) и фагоцитов. Это обеспечивает беспрепятственное размножение бактерий, которое на 12 ч и более опережает массированный выход нейтрофилов из циркуляции. Благодаря воспалительной реакции цереброспинальная жидкость обогащается белками плазмы, в том числе опсонинами. Бактерии подвергаются опсонофагоцитарной атаке, которая, запаздывая, не всегда обеспечивает выздоровление.
Попытки найти специализированные факторы, способствующие закреплению менингококков на мозговых оболочках, не увенчались успехом. Известно лишь о перестройке цитоскелетного аппарата эндотелиальных клеток и их возможных, в том числе бактериальных, механизмах. Дело, по-видимому, ограничивается контактной болезнетворностью для эндотелиоцитов (хотя геном N. meningitidis не содержит генов, кодирующих систему секреции типа III) и трансцитозом, прежде всего в зоне хориоидного сплетения. Инвазию, как в случае с эпителием, скорее всего начинают бактериальные пили и CD46 эндотелиоцитов. После редукции пилей к реакции подключаются белки наружной мембраны и, возможно, другие факторы, в частности бактериальная IgA-протеаза, содействующая внутриклеточному выживанию менингококка. Впрочем, подобные поиски, возможно, излишни, так как инфицированные тромбы, образующиеся в сосудах, сами по себе — отличный субстрат для бактериальной колонизации.
Выявление грамотрицательных диплококков при микроскопии мазка из спинномозговой жидкости служит стопроцентным подтверждением диагноза менингококкового менингита (рис. 5).

Бактерии лежат свободно или находятся внутри нейтрофилов. Помогает выявление менингококковых антигенов в реакции агглютинации микрочастиц, нагруженных антименингококковыми антителами. Менингококк может быть выделен из крови, спинномозговой жидкости, кожных экзантем, носоглотки. В последнем случае его приходится дифференцировать от непатогенных нейссерий, постоянно обитающих в верхних дыхательных путях.
Утверждение представлений о протективности противокапсульных антител явилось основой для разработки вакцин из очищенных капсульных полисахаридов наиболее распространенных серогрупп менингококка (А, В, С). Однако их внедрение столкнулось с рядом трудностей. Полисахарид В, являясь полимером сиаловой кислоты, оказался слабым иммуногеном для человека, а полисахарид С лишен протективного эффекта у детей до двух лет (возможно, по причине Т-независимости), хотя именно в этом возрасте около 30% заболеваний связано с данной разновидностью менингококка. Предложены химические модификации В-полисахарида и его конъюгаты с белковыми носителями, стимулирующие синтез протективных антител, но, в целом, высокоэффективная вакцинация остается делом будущего. Есть предложение использовать с этой целью поверхностные трансферинсвязывающие белки, которые индуцируются во время инфекции. Антитела к трансфериновым рецепторам (прежде всего к Tbp2) блокируют связывание трансферина, обладая бактерицидностью. Принципиально, что по антигенной специфичности трансферинсвязывающие белки одинаковы у большинства штаммов менингококка. В перспективе это определяет широкий спектр вакцинального эффекта. Впрочем, учитывая невысокую заболеваемость и ее редкие подъемы, введение плановых прививок вряд ли целесообразно. Их следует проводить скорее по эпидемическим показаниям, т.е. в период вспышек. Полезно помнить и о том, что сывороточные антитела, защищающие от внутрисосудистой инвазии, не предохраняют от носительства менингококка, а потому не способны обеспечить искоренения инфекции.
Во многих случаях спасает то, что менингококки сохраняют чувствительность к пенициллину, современные разновидности которого, наряду с новыми поколениями цефалоспоринов, остаются лучшим средством этиотропной терапии. Хотя и с большими потерями, антибиотики проходят через гематоэнцефалический барьер, достигая бактерицидных концентраций в цереброспинальной жидкости. Этому способствует повышение проницаемости сосудистых сплетений головного мозга на фоне воспаления — именно через них идет формирование основной массы тканевой жидкости и вместе с ней поступление в ЦНС лекарственных препаратов. Массовый бактериолиз, индуцируемый антибиотиками, может спровоцировать высвобождение больших количеств эндотоксина. Этим объясняется временное ухудшение состояния некоторых больных после первых инъекций антибиотиков. Поэтому в целях безопасности их применяют одновременно с кортикостероидными гормонами, которые сглаживают действие флогогенных цитокинов, опосредующих септические эффекты эндотоксина.
Гонококк
Мала причина, да грех велик.
- Особенности микробиологии.
- Эпидемиология мужской и женской гонореи.
- Адгезины и колонизация эпителия.
- Инвазивность и пиогенность.
- Острая гонорея и патогенез осложнений.
- Механизмы персистенции и хронической гонореи.
- Проблемы лечения и профилактики.
Г онорея — одна из самых распространенных инфекций человека, передаваемых половым путем. В переводе с греческого гонорея означает истечение семени. Название болезни, предложенное Галеном за 130 лет до н.э., с патогенетической точки зрения ошибочно (истекает не семенная жидкость, а гной), но отлично отражает один из главных симптомов острой инфекции у мужчин — обильное выделение гнойного экссудата из мочеиспускательного канала при остром гонорейном уретрите. Еще образнее немецкое наименование болезни — триппер (от нем. trippen — капать). Оба термина символизируют мужскую гонорею; у женщин заболевание протекает менее остро, нередко в скрытой форме. Впрочем, это не делает женскую гонорею менее опасной. Инфекция обретает коварный характер, создавая опасность осложнений и затрудняя контроль за распространением.
Острая гонорея может переходить в хроническую форму, которую непросто диагностировать и трудно лечить. Вялотекущее гнойное воспаление распространяется на различные отделы полового тракта, соседние ткани и имеет тяжелые последствия, связанные с реактивным разрастанием фиброзной ткани — склерозом. Изредка гонорея осложняется септикопиемией с преимущественным поражением суставов, клапанов сердца, аорты, кожи. Яркой манифестацией экстрагенитальной гонореи является гнойный конъюнктивит, или бленорея (от греч. blennos — слизь и rheo — теку). Кроме взрослых, бленореей болеют новорожденные, заражаясь в родовом канале матерей, инфицированных гонококком.
Возбудитель открыт в 1879 г. А. Нейссером. Он сообщил о диплококках, в большинстве располагающихся внутриклеточно, обнаруженных им у всех больных с острым уретритом и бленорейным воспалением глаз (рис. 1). А. Нейссер предложил называть вновь открытые бактерии гонококком или гонорейным диплококком. Вскоре были получены чистые культуры гонококка и при заражении ими добровольцев воспроизведена гонорея (гонорейный уретрит). Это окончательно утвердило за диплококком Нейссера роль возбудителя гонореи. Гонококк является строгим паразитом человека, хотя в эксперименте гонорею удается вызвать у приматов, в частности шимпанзе. Они не только «болеют» гонореей, но и передают ее половым партнерам.

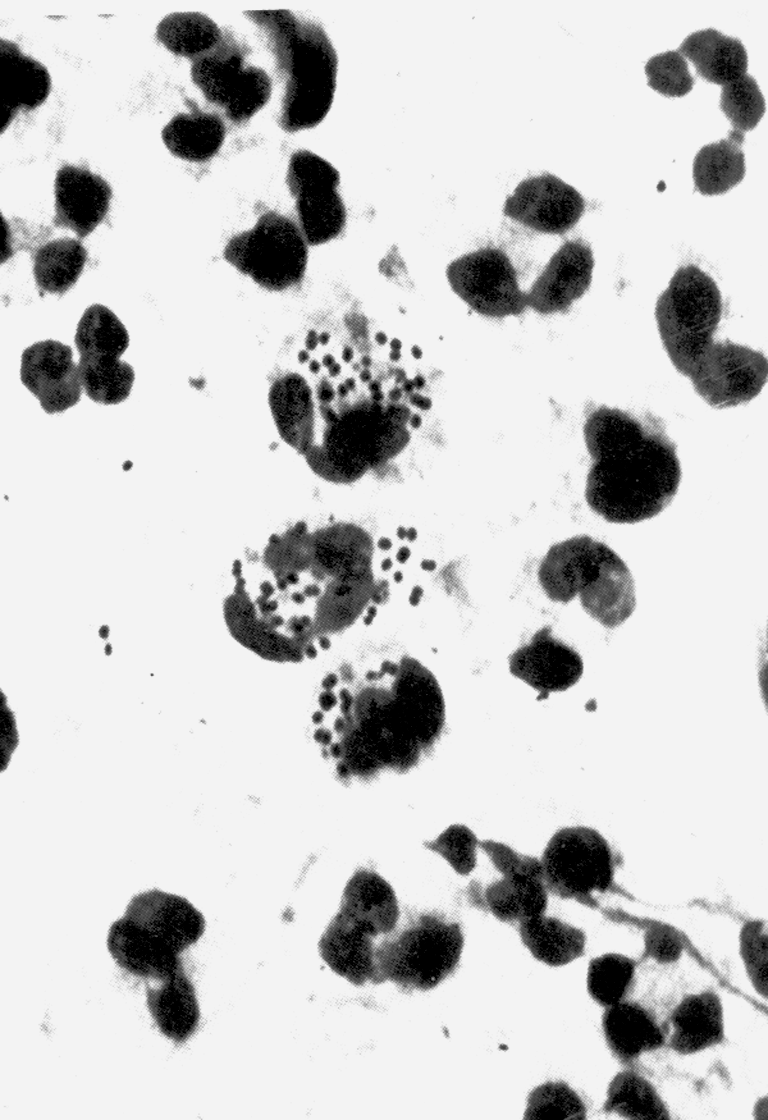
В 1933 г. бактерии, родственные гонококку, были объединены в семейство Neisseriaceae, а гонококк включен в один из его родовых таксонов (Neisseria) в качестве вида, Neisseria gonorrhoeae. Гонококк и менингококк (Neisseria meningitidis) — единственные патогенные виды этой небольшой группы грамотрицательных диплококков. Они обладают высокой степенью генетического родства (более 90% гомологии ДНК) и с точки зрения чистой генетики могут рассматриваться как подвиды (эковары) одного вида. Однако практические соображения (клиническая дискретность гонококковой и менингококковой патологии) сдерживают академический формализм: менингококк и гонококк продолжают считать разными видами. Что касается остальных нейссерий, то они относятся к нормальным обитателям слизистых оболочек ротовой полости, верхних дыхательных путей и мочеполового тракта человека.
Гонококки неподвижны, не образуют спор, в свежих культурах имеют микрокапсулу, которая в отличие от менингококка быстро исчезает и не участвует во взаимоотношениях с хозяином. Подобно менингококку, большинство клеток собраны в пары и обращены друг к другу уплощенной поверхностью — вид кофейного боба (рис. 2).

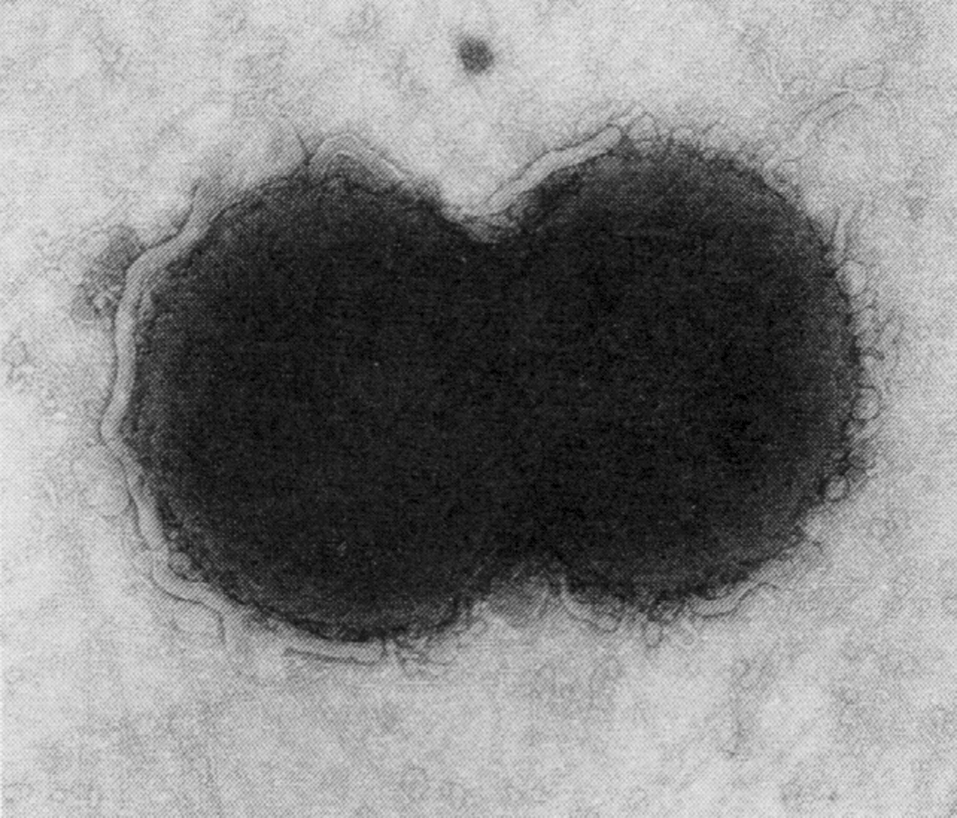
Гонококк принадлежит к наиболее прихотливым бактериям. Он требует сложных питательных сред, капнофилен (лучше растет в атмосфере 3—10% СО2), не устойчив во внешней среде (отсюда малая вероятность заражения путем непрямого контакта, например через предметы обихода), склонен менять культуральные, ферментативные и антигенные свойства как в ходе инфекционного процесса, так и при пассировании на питательных средах. Это создает немало проблем при выделении первичных культур, их сохранении в исходном состоянии, исключает возможность серотипирования штаммов и пр. Большое значение имеет способность усваивать железо. Для этого в наружной мембране гонококка есть белковые рецепторы (сходные с N. meningitidis), извлекающие железо из трансферина, лактоферина, гемоглобина и сидерофоров других бактерий. Наибольший функциональный смысл имеют трансфериновые рецепторы, ТbrA и TbrB. Они реагируют только с трансферином человека — одна из причин того, что гонококк является сугубо человеческим патогеном. Мутанты, не обладающие способностью связывать трансферин, лишены вирулентности. Механизм действия TbrA/TbrB объясняется конформационными изменениями трансферина, которые ведут к высвобождению фиксированного железа и его транспорту в периплазматическое пространство.
Паразитирование на человеке является обязательным для выживания гонококка. Он достигает этого благодаря 1) распространению путем наиболее эффективного (с точки зрения микробной контаминации) прямого контакта, изобретенного природой, 2) внутриклеточному паразитированию, 3) антифагоцитарным свойствам, 4) ускользанию от эффекторов иммунитета (антител), 5) способности к адаптивной устойчивости против антибиотиков. Представления о механизмах, обеспечивающих эти признаки, являются ключом к патогенетической трактовке гонорейного процесса.
В основе острой гонореи лежит поражение эпителия, и в этом смысле ее можно рассматривать как эпителиальную инфекцию. Обычно процесс начинается с ворсинок цилиндрического эпителия слизистой оболочки мочеполового тракта (в редких случаях в процесс вовлекаются верхние дыхательные пути). У мужчин первично поражается передняя уретра, у женщин — чаще цервикальный канал. Мужская гонорея — почти всегда клинически выраженный процесс с обильными гнойными выделениями (гнойный уретрит). Для женской гонореи характерно менее острое течение; в 60—80% случаев болезнь развивается скрытно, заявляя о себе при заражении половых партнеров. Женщины могут оставаться клинически латентными носителями недели и месяцы, что создает большие трудности для слежения за циркуляцией возбудителя.
При половых контактах с гонорейным партнером инфицируется около половины женщин; для мужчин риск заражения составляет 25—50%.
Поэтапная инвазия гонококков сквозь многослойный эпителий в субэпителиальную ткань вызывает интенсивную воспалительную реакцию с образованием гнойного экссудата. Это ответ на функциональное и структурное повреждение эпителия, которое воспринимается как сигнал к мобилизации фагоцитов крови (нейтрофилов). Контактным путем (по продолжению) и по лимфатическим сосудам инфекция распространяется на соседние ткани. У мужчин в процесс вовлекаются железы Литтре, предстательная железа, семенные пузырьки, куперовы железы, семявыносящий проток и придатки яичек. У женщин серьезные проблемы связаны с восходящей инфекцией органов малого таза (поражение эндометрия, фаллопиевых труб, яичников, брюшины). Показательно, что влагалище, покрытое многослойным плоским эпителием, не поражается гонококком. Это объясняется кислой средой, создаваемой лактобактериями вагинальной микрофлоры. Они утилизируют гликоген, которым богаты эпителиоциты влагалища взрослой женщины, сбраживающие его до молочной кислоты. У девочек гонококк охотно колонизирует наружные половые органы и влагалище, вызывая вульвовагинит. Смена вагинальной экологии (рН), наступающая с половой зрелостью, обычно полностью излечивает от этой формы гонореи.
Острый процесс не всегда завершается удалением возбудителя. Гонококк склонен к персистенции, которая в отличие от ряда других патогенных бактерий не является бессимптомным носительством на фоне приобретенного иммунитета, но отражает переход в иную, хроническую, фазу патологического процесса. В известном смысле это результат коадаптации хозяина и паразита, благодаря которой возбудитель обретает возможность дождаться новой среды обитания, т.е. проинфицировать другой организм и найти нового хозяина. Если во внешней среде гонококк быстро погибает, то в зараженном организме без вмешательства антибактериальных препаратов он с трудом поддается уничтожению. Инициируя процесс, гонококк нередко прокладывает дорогу другим возбудителям, так что хронизация часто эволюционирует как смешанная патология, в которой значительную роль играет негонококковая инфекция. Поэтому нередко приходится говорить уже не о гонорее, а о постгонорейных осложнениях. В таких случаях гонококк не выделяется, но процесс поддерживается за счет вторичной (чаще хламидийной) инфекции. Реактивное разрастание фиброзной соединительной ткани ведет к образованию спаек. Кульминацией являются стриктуры уретры, фаллопиевых труб, семявыносящего протока и, как следствие, бесплодие, внематочная беременность и пр. При малосимптомной женской гонорее это воспринимается как неожиданность.
Сравнительно редко гонококк проникает в кровяное русло, давая начало генерализованной инфекции и экстрагенитальным осложнениям. Для гонококковой бактериемии характерны гнойные поражения суставов и кожи, хотя выделить возбудитель из очагов инфекции удается не всегда. Иногда развивается эндокардит, менингит. Клинические признаки гонококцемии могут наблюдаться при бессимптомном местном (генитальном) процессе, особенно у женщин.
К драматичным проявлениям гонококковой инфекции относится острый гнойный конъюнктивит (бленорея) новорожденных. Заражение происходит от инфицированной матери внутриутробно, во время родов или после рождения. Профилактический эффект достигается закапыванием в глаза 1% раствора нитрата серебра или капель из антибиотиков. Бесконтрольное развитие процесса грозит слепотой. Аналогичные поражения конъюнктивы встречаются и у взрослых.
Гонорея принадлежит к числу наиболее заразных болезней человека. Это во многом связано с циркуляцией высоковирулентных штаммов гонококка, которые инициируют патологический процесс при инстилляции в уретру всего 1000 клеток. Являясь классической пиогенной инвазией слизистых оболочек, гонорея начинается с колонизации эпителиоцитов. В закреплении бактерий на поверхности эпителия участвует комплекс поверхностных компонентов (адгезинов) гонококка. Наиболее очевидна роль пилей. Все штаммы, выделяемые от больных острой гонореей, обладают пилями (рис. 5), и только такими культурами удается заразить добровольцев.
Пронизывая капсулу, пили связываются с сиалированными рецепторами (СD46) многих клеток, включая эритроциты, эпителиоциты слизистой оболочки мочеполового тракта, буккальные эпителиоциты, сперматозоиды. Белок пилей почти всех штаммов гонококка отличается разнообразием, так что каждый из них содержит дискретные антигенные формы пилина. Кроме чисто механического эффекта (предупреждение вымывания бактерий мочой), пили способствуют внутриэпителиальной инвазии — патогенетически значимому этапу гонококкового процесса. Адгезия на сперматозоидах (рис. 6) создает дополнительную возможность для проникновения гонококка в верхние отделы женского полового тракта.



На протяжении нескольких лет пилезависимая адгезия воспринималась как главный механизм гонококковой инфекции, через который удастся управлять специфической профилактикой гонореи и контролировать патологический процесс. Но вопрос оказался сложнее: адгезия, детерминируемая пилями, — лишь одна из причин мукозальной колонизации. Достаточно сказать, что пилями обладают невирулентные штаммы гонококка и непатогенные нейссерии. Это побуждает искать иные (точнее, дополнительные) механизмы инициации гонорейного процесса.
Привлекают внимание белки наружной мембраны, PorA/PorB (pI), Opa (pII) и Rmp (pIII). Они заметно влияют на взаимоотношения с хозяином, усиливая адгезию гонококка, внутриклеточную инвазию, антифагоцитарную активность, устойчивость к бактерицидным факторам сыворотки. Каждый штамм экспрессирует только один тип Por (порины PorA или PorB), хотя всего известно около 20—30 сероваров PorA и PorB. Наряду с липополисахаридом (липоолигосахаридом — см. «Менингококк»), Por служит мишенью для бактерицидных антител, но может избегать их благодаря выраженному эпитопному своеобразию, а также блокирующему действию белка Rmp (см. «Менингококк»).
Opa-протеины называются так потому, что их присутствие (Ора+) делает колонии непрозрачными из-за аггрегации бактерий (англ. opaque — непрозрачный); прозрачные колонии (Ора—) лишены Ора-белков. Ора-зависимая агрегация, возможно, усиливает контакт между бактериями, содействуя обмену генетической информацией, типичному для нейссерий. Каждый штамм имеет более десятка аллельных ора-генов для А—К-белков, но одновременно экспрессирует один-два, редко больше, причем в клональных вариантах. Это связано со способностью гонококка менять свои антигены (см. ниже). Ора-белки включаются в адгезию, необходимую для полноценного (интимного) взаимодействия с эпителиальными клетками, эндотелиоцитами и нейтрофилами. Речь идет о реакциях, заканчивающихся трансцитозом (инвазией) бактерий.
Принято считать, что для инвазии необходимо последовательное участие пилей и белков наружной мембраны. После пилезависимого подключения Ора-белков возникает прямая или опосредованная (витронектин/фибронектинзависимая) рецепция с гепаринсульфатпротеогликанами и/или молекулами СD66. Результатом является инвазия бактерий и продукция флогогенных цитокинов/хемокинов, запускающих воспаление. Культуры гонококка обычно представляют смесь непрозрачных и прозрачных колоний, первые доминируют при уретрите у мужчин и неосложненной гонорее у женщин. Прозрачные (особенно пилепозитивные) варианты устойчивее к бактерицидному действию сыворотки и фагоцитозу, доминируя при генерализованных формах гонореи. Точные причины этого подлежат выяснению.
Впрочем, несмотря на логику, роль Ора-белков остается неясной. Дело в том, что гепаринсульфатные протеогликаны расположены на базолатеральной поверхности эпителия и в норме не доступны для гонококка. Поэтому Ора-белки не могут быть пусковым фактором инвазивной адгезии. Они способны усилить уже начавшийся процесс, инициируемый воспалением, которое повышает проницаемость мукозального барьера. Показано, что для этого гонококку необходимы пили и, возможно, ряд других компонентов, таких как PorA/PorВ и липополисахарид (липоолигосахарид).
Подобно другим паразитам слизистых оболочек, гонококк продуцирует IgA-протеазу. До недавних пор считалось, что ее единственной функцией является разрушение IgA1 и связанное с этим вмешательство в антиадгезивную активность антител на поверхности эпителия. Оказалось, что это не так. IgA-протеаза разрушает внутриклеточный протеин LAMP-1 (он имеет структурное сходство с шарнирным участком IgA1), что препятствует образованию фаголизосом, помогая выживанию нейссерий внутри эпителиальных клеток.
Проходя сквозь многослойный эпителий и пенетрируя его базальную мембрану, гонококки проникают в субэпителиальную ткань, возбуждая острую воспалительную реакцию. Механизм деструкции эпителия при гонорее не ясен. Кроме слабого гемолизина, гонококк не секретирует никаких токсических факторов, и за неимением лучшего повреждение слизистых оболочек связывают с липополисахаридом (эндотоксином) наружной мембраны. Он выделяется в виде каплевидных образований (англ. blebs), представляющих смесь эндотоксина и белков наружной мембраны. Это происходит при избыточной продукции компонентов клеточной стенки, прежде всего в молодых гонококковых культурах.
Цитотоксическими свойствами обладают производные гонококкового пептидогликана. При введении в уретру или в полость сустава они обнаруживают пиогенную активность и вызывают деструкцию слизистого покрова в органных культурах фаллопиевых труб. Впрочем, не исключено, что микробные факторы играют лишь роль пускового стимула. Основную альтерацию вызывают продукты активированных нейтрофилов, которые невольно включаются в деструктивный процесс. В этом диалектика воспалительных реакций, сочетающих в себе элементы защиты и повреждения.
Высокая пиогенность отражает, как минимум, еще один признак — относительную устойчивость гонококка к бактерицидному действию нейтрофилов. Низкий коэффициент полезного действия фагоцитарной реакции компенсируется за счет ее количественного усиления, т.е. повышенной эмиграции нейтрофилов в зону инфекции. Гнойные клетки (так со времен Нейссера называют нейтрофилы, нафаршированные бактериями) служат символом и надежным диагностическим критерием гонореи (см. рис. 1). Издавна сложилось представление о незавершенном фагоцитозе гонококка, т.е. о неспособности нейтрофилов переваривать поглощенные бактерии. В действительности картина выглядит иначе. Большинство гонококков погибает при фагоцитозе, но те, что выживают, дают начало клонам с повышенным антифагоцитарным потенциалом. В отличие от менингококка, гонококки лишены капсулы, влияющей на взаимоотношения с фагоцитами. Более активное сопротивление фагоцитозу оказывают пили гонококка: штаммы, утратившие пили, менее устойчивы к поглощению и быстрее уничтожаются нейтрофилами. При угасании заболевания и при переходе в хроническую форму большинство гонококков располагается свободно на поверхности эпителия, ускользая от фагоцитарной атаки. Возможно, это связано с отбором клонов, устойчивых к фагоцитозу. Как уже говорилось, в адсорбции гонококка на нейтрофилах участвуют белки наружной мембраны: бактерии, дефектные по Ора-протеинам, фагоцитируются слабее. Избежать встречи с фагоцитами помогает и внутриэпителиальная инвазия, благодаря которой гонококк получает временное убежище от нейтрофилов.
С серьезными проблемами гонококк сталкивается в кровяном русле. Сыворотка лизирует его (как и большинство других грамотрицательных бактерий) при помощи комплемента, который активируется по антителонезависимому (лектиновому/альтернативному) пути или в содружестве с IgG- и IgM-антителами (классический путь). Штаммы, выделяемые при генерализованной инфекции, отличаются повышенной устойчивостью к бактерицидному действию сыворотки. Это коррелирует с сокращением спектра наружных антигенов, которые могут быть атакованы антителами, и появлением в составе мембранного липополисахарида сиаловой кислоты, блокирующей активацию лектинового/альтернативного каскада комплемента и сборку цитолитического комплекса (С5—С9). Дефект по терминальным факторам комплемента повышает вероятность системных гонококковых осложнений.
Патогенетически важным свойством гонококка является изменчивость его поверхностных антигенов, от которых зависит приобретение протективного иммунитета. Она формируется на основе полиморфизма генов и обеспечивает появление клонов с новыми антигенными эпитопами, не распознаваемыми ранее образовавшимися антителами. В структуре пилей и белков наружной мембраны (прежде всего Ора) имеются гипервариабельные участки, кодируемые супермутабельными и/или мозаичными генами. Последние собираются из территориально разобщенных фрагментов ДНК и в результате их случайных сочетаний вносят обновленную информацию о структуре белковых антигенов. Генетический механизм, детерминирующий антигенное разнообразие гонококков (он действует и у ряда других бактерий), напоминает стратегию формирования гипервариабельных участков иммуноглобулинов, определяя клональную специфичность антигенов, состоящую из множества антигенных типов и субтипов гонококка.
Опережающая перестройка поверхностных компонентов способствует персистенции гонококка, создавая возможность селекции клонов, которые инертны к антителам и лучше закрепляются в новых биотопах благодаря структурным вариантам адгезинов с повышенным сродством к рецепторам эпителиоцитов. Это создает предпосылки для хронической гонореи и ее различных (генитальных, парагенитальных и экстрагенитальных) осложнений. Этим же объясняются неудачные попытки серологических классификаций гонококка и сомнительные перспективы создания эффективной гонококковой вакцины.
Гонорейный процесс сопровождается местной и системной иммунной реакцией. В секретах и воспалительном экссудате полового тракта появляются IgA- и IgG-антитела, которые блокируют адгезию гонококков на эпителиоцитах и усиливают фагоцитоз опсонизированных бактерий. В крови накапливаются IgG- и IgM-антитела с комплементзависимой бактерицидностью. Но эффективность приобретаемого иммунитета невелика. Человек может многократно болеть гонореей и даже во время не вполне излеченного или хронического процесса заражаться повторно. Противоречие между выраженными иммунными сдвигами и отсутствием резистентности логично вписывается в представления об антигенной вариабельности гонококка. Складывается подобие порочного круга: хозяин адаптирует свой иммунитет к инфицирующему штамму, но одновременно способствует селекции бактерий, ускользающих от иммунологического надзора благодаря новым антигенным свойствам. Этому содействуют и так называемые фазовые вариации — прекращение синтеза ряда антигенов, в частности пилей и Ора-белков внешней мембраны. Это отражается на культуральных свойствах гонококка (тип колоний) — феномене, который известен давно и сопряжен с вирулентностью возбудителя. Лучше всего он проявляется на плотных питательных средах, где гонококк дает колонии четырех—пяти типов. Вирулентностью обладают типы 1 и 2, и только они обладают пилями (см. рис. 5).
В целом клинические наблюдения и эксперименты на животных склоняют к нестандартному выводу: иммунитет при гонорее носит клональный (штаммовый) характер, т.е. организм получает защиту против «своего» гонококка, но сохраняет чувствительность к «чужим» штаммам. Впрочем, устойчивость к собственному штамму тоже относительна, и больной может вновь пережить острую гонорею при уретральном заражении гонококком, находившимся до этого времени в латентном состоянии в других тканях, например в предстательной железе. Весьма вероятно, что в подобных случаях рецидивы острой инфекции связаны с антигенными вариантами гонококка, избегающими антител против исходного штамма. Это объясняет ряд удивительных фактов, которыми богата история гонореи. К примеру, больной хронической гонореей, заражая здоровую женщину своими гонококками, может при последующих половых контактах остро инфицироваться от нее собственными бактериями, но уже подвергшимися пассажу через другой организм. В таких случаях мужчина нередко обвиняет женщину в своем заражении, хотя она играет здесь лишь роль передатчика его же собственных гонококков, претерпевших адаптивную перестройку в новой для себя среде. С высокой антигенной вариабельностью гонококка связаны и неудачи специфической вакцинопрофилактики и вакцинотерапии гонореи.
Антитела к гонококковым антигенам можно обнаружить при помощи различных серологических реакций. Их диагностическая ценность при острой гонорее невелика, но они полезны в дифференциальной диагностике хронических генитальных инфекций. При острой гонорее у мужчин микроскопия мазка из уретры практически гарантирует диагноз. В нем обнаруживаются «гнойные клетки», т.е. нейтрофилы с поглощенными диплококками (см. рис. 1). При женской и хронической гонорее наиболее надежным остается выделение и идентификация чистой культуры. Широко используется экспресс-диагностика, основанная на иммунологическом выявлении микробных клеток и их антигенов в патологическом материале. Возлагаются надежды и на генетические маркеры гонококковой инфекции. Коммерческие наборы для полимеразной цепной реакции уже рекомендованы к практическому применению.
Долгое время почти абсолютным средством против гонококков был пенициллин. Однако постепенно многие штаммы потеряли к нему чувствительность прежде всего из-за распространения плазмид, кодирующих способность к разрушению пенициллинов. Поэтому сегодня используют цефалоспорины, устойчивые к гонококковой бета-лактамазе, а также новое поколение макролидов, например азитромицин. Они быстро обрывают острый процесс, но это не всегда сочетается с избавлением от возбудителя. Необходимо внимательно обследовать больного для исключения остаточного инфекта, грозящего переходом в хроническую гонорею.
Палочка инфлюэнцы
Я так зовусь, хоть имя не мое.
- Ошибка видового эпитета.
- Природа гемофильности.
- Капсульные серовары.
- Капсула типа b: главный фактор пиогенной инвазии.
- Место в тройке лидеров возбудителей гнойного менингита.
- Трагический финал местных инвазий.
- Бескапсульные штаммы и патология респираторного тракта.
- Т-независимость капсульных антигенов.
- Конъюгированные вакцины.
Палочка инфлюэнцы (Haemophilus influenzae) стала известна в 1889—1893 гг. Первенство открытия делят микробиологи России (М.И. Афанасьев), Германии (Р. Пфейфер) и Японии (С. Китазато). Их объединяла общая цель — найти возбудителя гриппа, который иначе назывался инфлюэнцей (о происхождении термина см. «Вирусы гриппа»). Именно в носоглотке больных гриппом микроб был обнаружен путем бактериоскопии, а затем выделен в чистой культуре из мокроты. Иллюзия о бактериальной этиологии гриппа продержалась почти 25 лет, до самой страшной его пандемии — «испанки». Она унесла больше жизней, чем первая мировая война и возродила интерес к изучению природы этого заболевания. Что касается палочки инфлюэнцы (ее тогда называли палочкой Пфейфера, так как именно Пфейфер особенно упорно отстаивал значение H.influenzae в происхождении гриппа), то оказалось, что она с одинаковой частотой выделяется от больных гриппом и здоровых людей. Это означало, что ее нельзя считать причиной гриппа, по крайней мере его основным возбудителем. Вопрос был окончательно решен в 1933 г., когда удалось доказать контагиозность безбактериального фильтрата мокроты гриппозных больных, а затем изолировать вирус(ы) гриппа.
Интерес к палочке Пфейфера упал. В сочетании с трудностями ее выделения и культивирования это привело к тому, что она до сих пор нередко остается в тени практической бактериологии. Между тем, по данным мировой статистики, палочка инфлюэнцы занимает одно из первых мест среди факторов детской смертности. Она особенно опасна для детей до 5 лет, являясь наряду с пневмококком наиболее вероятной причиной острых гнойных поражений среднего уха (средний отит), придаточных пазух носа (синуситы), верхних дыхательных путей (ларинготрахеит, эпиглоттит) и мозговых оболочек (здесь H. influenzaе делит «лавры» с пневмококком и менингококком). Но палочка инфлюэнцы не безопасна и для взрослых, осложняя вирусные (грипп) и бактериальные (коклюш) инфекции респираторного тракта. Она служит причиной вторичных бронхопневмоний, поддерживая их хроническое течение и вызывая обострения.
Присутствие палочки инфлюэнцы в носовой полости и носоглотке большинства здоровых людей (начиная с 3 мес контаминированы практически все дети) вынуждает рассматривать ее как условно-патогенные бактерии, которые при благоприятном для себя стечении обстоятельств нарушают «перемирие» с хозяином. Впрочем, подобная логика для палочки инфлюэнцы слишком примитивна. Ее штаммы сильно различаются по вирулентности, наиболее ярким маркером которой служит капсула. Все проблемы острой инвазивности (бактериемия, осложненная менингитом) связаны со штаммами, образующими капсулу типа b. Есть даже мнение, что палочку инфлюэнцы требуется расчленить по меньшей мере на два вида, один из которых следует закрепить за типом b. Другие разновидности ограничивают свои экологические претензии колонизацией респираторного тракта, а попадая в кровь, быстро погибают. Поэтому, говоря о палочке инфлюэнцы как о почти облигатном симбионте человека, необходимо помнить, что речь идет не о всех ее разновидностях. Некоторые из них, прежде всего обладающие капсулой b, редко выделяются от здоровых и ближе стоят к первичным патогенам. Более того, даже штаммы типа b неоднородны: известно более 100 клонных вариантов, из которых только 6 вызывают 80% инвазивных инфекций.
Однако и бескапсульные формы H. influenzae представляют немалый интерес для медицины. Персистируя за счет закрепления на муцине носоглоточной слизи, они нередко служат причиной среднего отита и синуситов. С ними связана широко распространенная патология респираторного тракта — хронический бронхит. Этому способствует мутационная изменчивость иммунодоминантных белков наружной мембраны. Она обеспечивает селекцию и длительное выживание бактерий на фоне иммунного ответа.
Современное название палочки инфлюэнцы (Haemophilus influenzae) сочетает в себе память об искренних заблуждениях ученых, считавших ее возбудителем гриппа (инфлюэнцы), и абсолютную зависимость от крови при росте на искусственных питательных средах. Родовое наименование (Haemophilus) распространяется на небольшую группу бактерий (около 20 видов), причисляемых к так называемым гемофилам. Их уникальная зависимость от крови обусловлена тем, что, предпочитая аэробный тип метаболизма, они не способны синтезировать гемин, входящий в состав ферментов дыхательной цепи, и никотинамидадениндинуклеотид — НАД (или НАДФ) — ключевой кофактор окислительно-восстановительных ферментов. Одного этого вполне достаточно, чтобы объяснить абсолютную зависимость гемофилов от человека и животных. Оба ростовых фактора (гемин и НАД — факторы Х и V) эти бактерии легко получают из крови, а потому их всегда культивируют на кровяных средах. Лучше пользоваться «шоколадным» агаром (он имеет темно-коричневый оттенок), который получают прогреванием кровяного агара при 80°С для высвобождения из эритроцитов гемина и НАД.
Если кровь (эритроциты) является единственным источником гемина, то обогащения среды фактором V можно добиться разными способами. При изготовлении питательных сред его стандартным источником служит экстракт дрожжей. Требуется лишь помнить, что в отличие от гемина НАД не очень устойчив к высокой температуре и длительное кипячение (с целью экстрагирования) разрушает его. На зависимости от НАД построен остроумный способ индикации H. influenzae: его колонии вырастают рядом с бактериями (например, стафилококком), продуцирующими НАД (см. рисунок). Разумеется, что в этом случае необходимо использовать питательные среды, содержащие только один из ростовых факторов — гемин. Этого легко добиться, если кровяную добавку подвергнуть жесткой термической обработке, например, автоклавированию при 120°С.

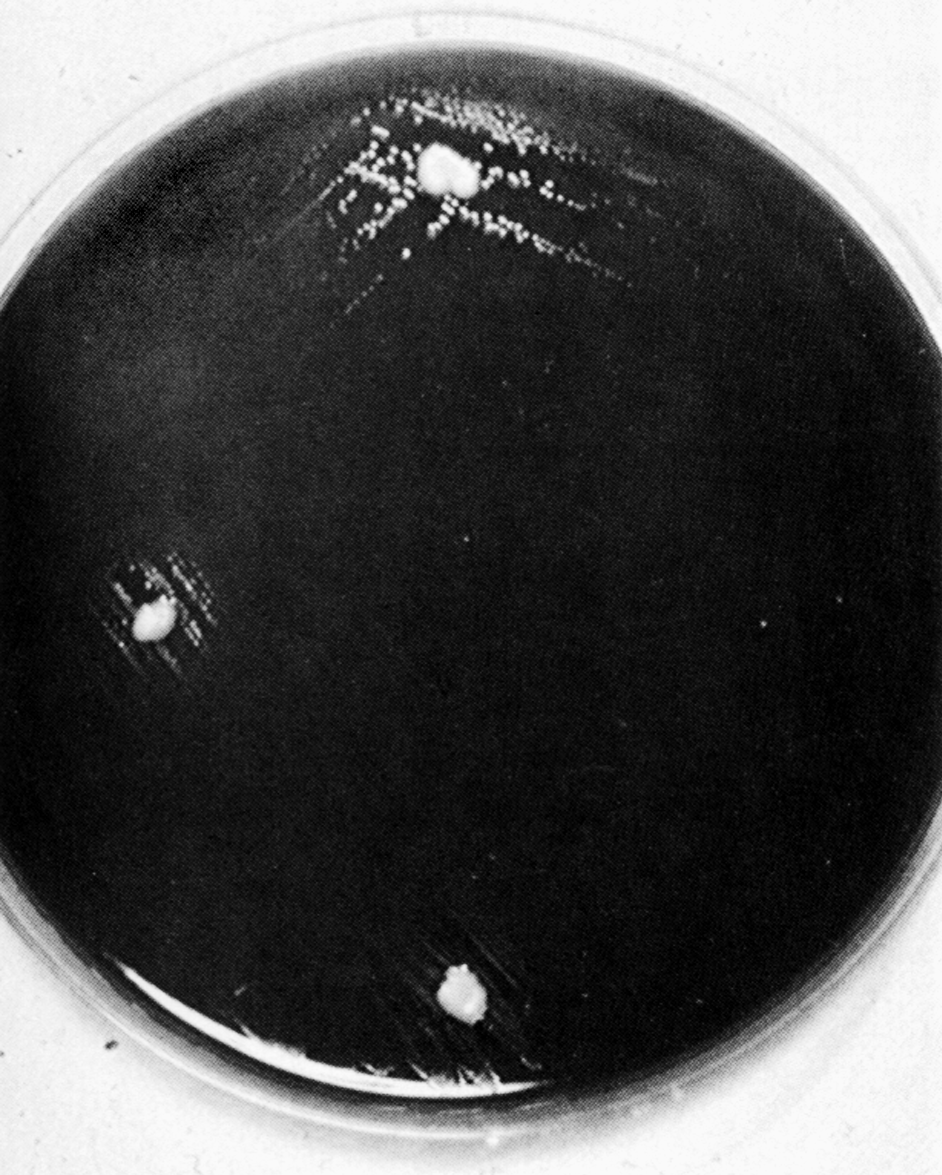
продуцирующего фактор V/НАД
Кроме ауксотрофности (т.е. зависимости от ростовых факторов), характеристику H. influenzae как облигатного симбионта-паразита дополняет неустойчивость во внешней среде. Палочка инфлюэнцы быстро погибает вне организма, а потому распространяется лишь при непосредственных контактах путем воздушно-капельного распыления. Вся экология палочки инфлюэнцы связана с человеком, других ее резервуаров в природе не существует.
H. influenzae представляет собой мелкие (не более 1,5 мкм) палочки с закругленными концами. Характерно образование нитевидных (филаментозных) и других аномальных клеток. Эти бактерии грамотрицательны, спор не образуют, неподвижны, но, подобно всем грамотрицательным бактериям, имеют пили (фимбрии). Будучи факультативными анаэробами, лучше растут в аэробных условиях. Капсула (она построена из полисахаридов) является патогенетически важным, но не универсальным признаком. К тому же она выражена слабо и не заметна при обычной микроскопии. Чаще всего палочка инфлюэнцы живет на поверхности слизистой оболочки респираторного тракта и изредка выделяется из влагалища. Большинство штаммов, выделяемых от здоровых людей (колонизировано 50—80% населения) и из очагов местной инфекции (бронхит, пневмония, отит, синуситы), не имеют капсулы и ее антигенов (нетипируемые штаммы). Капсульные разновидности (они выделяются примерно от 5% здоровых людей) подразделяются на 6 типов: a, b, c, d, e, f. Дебатируется возможность приобретения или потери капсулы, в частности при S—R-диссоциации бактерий. Бескапсульные варианты рассматриваются как R-формы, которые при индукции зарепрессированных капсульных генов могут трансформироваться в капсульные S-варианты, обретая повышенную вирулентность. Впрочем, есть и другая версия, согласно которой капсульные и бескапсульные формы следует рассматривать как самостоятельные таксоны, не переходящие друг в друга. Об этом говорят существенные генетические различия фенотипически дискретных штаммов H.influenzae.
Капсула — главный атрибут инвазивности палочки инфлюэнцы. Она обеспечивает выживаемость бактерий в кровяном русле и дальнейшее развитие инфекционного процесса. Как уже говорилось, почти все случаи гнойного менингита, которым печально знаменита палочка инфлюэнцы, cвязаны c определенными b-клонотипами. Капсула b-типа представляет полимер рибозы и рибитола, скрепленных фосфорной кислотой. Подобно другим полисахаридам, построенным из однотипных, многократно повторяющихся блоков, b-капсульный полирибофосфат является Т-независимым антигеном, что накладывает существенный отпечаток на иммунитетные взаимоотношения с бактериями — носителями такого рода покрытий (см. ниже).
Из других факторов, имеющих отношение к патогенности, можно отметить липополисахаридный эндотоксин (он доминирует в развитии септического синдрома), пили (адгезия), IgA-протеазу (инактивация секреторных антител), фактор, подавляющий мукоцилиарный транспорт в системе реснитчатого эпителия (усиливает колонизацию). Никаких специфических токсинов у палочки инфлюэнцы не обнаружено, и вся ее болезнетворность связана, по-видимому, со способностью индуцировать пиогенное воспаление в зонах бактериальной инвазии — местных (например, в респираторном тракте) и/или гематогенных (менингит). В первом случае возбудитель пользуется «чужими» инфекционными и неинфекционными повреждениями слизистой оболочки, во втором — реализует свою агрессивность благодаря b-капсуле на фоне отсутствия антикапсульных антител.
Наиболее тяжелой патологией, связанной с палочкой инфлюэнцы, является гнойный менингит. В этом отношении с палочкой инфлюэнцы могут конкурировать лишь два вида бактерий — пневмококк и менингококк, причем в самой младшей возрастной группе (до 2—3 лет) H. influenzae претендует на абсолютное лидерство (см. таблицу).
Основные возбудители гнойного менингита
| Возраст | Бактерии | |
| Наиболее типичные | Прочие | |
| От рождения до 2 мес |
Str. agalactiae (группа В) | Е. coli Listeria monocytogenes |
| 2—60 мес | Н. influenzae (тип b) | N. meningitidis Str. pneumoniae |
| >60 мес | Str. рneumoniae | N. meningitidis* |
* Групповые заболевания – эпидемии.
Возрастная зависимость проявляется удивительно четко, и после 5 лет гемофильный менингит практически не встречается. Резистентны к нему и новорожденные до двухмесячного возраста, так как защищены материнскими антителами.
Антитела опсонизируют бактерии, проникшие в кровяное русло, обеспечивая их комплементзависимый лизис и удаление макрофагами ретикулоэндотелиальной системы. Эти механизмы работают и без антител, но они не эффективны против инкапсулированных бактерий. Поэтому патогенетически значимая инвазия (септическая бактериемия) всегда обусловлена капсульными штаммами H. influenzae. Это так же, как в случаях с менингококком и пневмококком, демонстрирует роль капсулы в устойчивости бактерий против факторов внутрисосудистого клиренса.
Остается лишь понять, почему среди всех капсульных вариантов палочки инфлюэнцы доминирует единственный — тип b. Здесь не обойтись без вполне конкретной иммунологии. Полирибофосфат, из которого построена эта капсула, относится к Т-независимым антигенам, антитела к которым образуются без участия Т-хелперов. Реактивность к таким антигенам отсутствует или снижена у младенцев, поэтому они лишены протективных антител. Кстати, примерно также обстоит дело с иммунитетом против пневмококка и менингококка, резистентность к которым быстро усиливается с возрастом, но эволюционирует неодинаково для разных капсульных сероваров. Например, антитела против пневмококкового полисахарида типа III появляются обычно к 6 мес, тогда как антитела против капсульного типа XIII обнаруживаются лишь у половины детей трехлетнего возраста.
Т-независимые антигены не оставляют иммунологической памяти, большинство антител против них относятся к иммуноглобулинам класса M. Последнее обстоятельство имеет немаловажное значение, так как в составе иммунных комплексов IgM-антитела отлично (лучше, чем IgG) активируют комплемент, запуская внутрисосудистый бактериолиз. Но они не проходят через плаценту, и лишь высокий уровень материнских IgG-антител обеспечивает пассивную защиту новорожденного от септической бактериемии. Соотношения между IgM и IgG антительным ответом не одинаковы для разных Т-независимых антигенов. Этим, в частности, можно объяснить особенности антиинвазивной врожденной защиты против палочки инфлюэнцы, менингококка и пневмококка. Усилить иммуногенность Т-независимых антигенов удается путем связывания с белковыми носителями, которые перерабатываются (процессируются) в макрофагах, представляющих антигены Т-хелперам. Это учитывается при изготовлении так называемых конъюгированных вакцин, предназначенных для профилактики гнойного менингита у детей. Но эволюция возрастной резистентности против H. influenzae типа b связана не только с созреванием аппарата реактивности к Т-независимым антигенам. Определенное значение имеют нормальные антитела, которые образуются при стимуляции антигенами нормальной микрофлоры, располагающими одинаковыми эпитопами с капсульным материалом палочки инфлюэнцы. Кроме того, следует учитывать и ограниченное распространение штаммов типа b. Человек с рождения колонизируется бескапсульными вариантами палочки инфлюэнцы и редко контактирует с ее капсульными антигенами.
Но эволюция возрастной резистентности против H. influenzae типа b связана не только с созреванием аппарата реактивности к Т-независимым антигенам. Определенное значение имеют нормальные антитела, которые образуются при стимуляции антигенами нормальной микрофлоры, располагающими одинаковыми эпитопами с капсульным материалом палочки инфлюэнцы. Кроме того, следует учитывать и ограниченное распространение штаммов типа b. Человек с рождения колонизируется бескапсульными вариантами палочки инфлюэнцы и редко контактирует с ее капсульными антигенами.
В опытах с мышатами установлено, что интраназальная инстилляция H. influenzae типа b является единственным условием для развития септической инвазии (системной инфекции). Более того, такие мыши легко инфицируют соседей по клетке. Около 4% детей в возрасте до 5 лет, находившихся в близком контакте с менингитным (H. influenzae) больным, заболевают гнойным менингитом. Возбудитель фиксируется на эпителии верхних дыхательных путей (очевидно, носа) и распространяется при распылении капель секрета слизистой оболочки. Механизмы, благодаря которым палочка инфлюэнцы прикрепляется к эпителию и преодолевает мукоцилиарный клиренс, точно не известны. Заметим лишь, что в отличие от бескапсульных штаммов капсульные формы не рецептируют муцинов (т.е. не задерживаются слоем слизи) и закрепляются на эпителиоцитах лишь после их удаления. Это означает, что свободные муцины секрета ослабляют адгезию инкапсулированных бактерий. Палочка инфлюэнцы инвазирует эпителиоциты и уже после этого проникает в субэпителиальную ткань. Благодаря капсуле бактерии ускользают от фагоцитоза и, проходя лимфоидный барьер, попадают в кровь.
Распространение бескапсульных вариантов сдерживается местной воспалительной реакцией. Она редко обретает острые формы, являясь одной из причин стабилизации хронической патологии бронхолегочного тракта у взрослых. Напротив, штаммы типа b чрезвычайно опасны и как возбудители местных пиогенных инвазий, самой грозной из которых является эпиглоттит — остро прогрессирующая инфекция (по существу флегмона) надгортанника и окружающих тканей. Эпиглоттит обычно поражает детей 2—5 лет на фоне назофарингита и может быстро привести к смерти от асфиксии. H. influenzae типа b — почти единственный возбудитель этой патологии, которая по значимости и распространению занимает второе место среди гемофильных инфекций.
Но вернемся к менингиту. При низком содержании противокапсульных антител бактерии выживают в крови, получая возможность инфицировать мозговые оболочки. Они достигают их через сосудистые сплетения мозга, где происходит образование спинномозговой жидкости. Поражение мозга является результатом острой пиогенной реакции, которая развивается в ответ на инвазию возбудителя. Воспалительный экссудат, скапливающийся в спинномозговом канале и желудочках мозга, служит отличной питательной средой для бактерий, обеспечивая их быстрое размножение. Блокада оттока цереброспинальной жидкости через арахноидальные ворсинки вызывает повышение внутричерепного давления и ранние признаки менингита — рвоту и летаргию. Васкулит и тромбофлебит мягкой мозговой оболочки ведут к ишемии и некротическим изменениям мозговой ткани. Невыразительные поначалу симптомы быстро нарастают так, что уже через 24—36 ч после заражения капсульным вариантом b развивается тяжелая патология, дающая (без лечения) до 90% летальных исходов.
В целом клиническая картина острого периода не отличается от таковой при менингококковом и пневмококковом менингитах. Подобно пневмококковому менингиту, гемофильный процесс чреват неврологическими осложнениями, которые зависят от места и степени поражения коры головного мозга. Даже при оптимальной терапии они возникают у каждого десятого больного, проявляясь в потере зрения, глухоте, обструктивной гидроцефалии, отставании умственного развития. Для пневмококка эта цифра еще выше — каждый третий из выживших детей. После менингококкового менингита неврологические осложнения наблюдаются редко. Одна из причин этих различий — неодинаковый масштаб пиогенного поражения субарахноидального пространства: оно незначительно для менингококка, умеренно при гемофильном менингите и ярко выражено при пневмококковой инфекции.
Этиологический диагноз быстро ставится путем микроскопического и иммунохимического анализов цереброспинальной жидкости. В последнем случае выявляют b-капсульный антиген в реакции со специфическими (анти-b) антителами, например путем агглютинации латексных частиц, покрытых анти-b. Если количество бактерий невелико, прибегают к бактериологическому исследованию.
H. influenzae чувствительны ко многим антибиотикам. Предпочтение отдается цефалоспоринам, хорошо проникающим в спинномозговую жидкость. Антибактериальная терапия существенно не влияет на вероятность летального исхода, но сглаживает течение болезни и снижает процент осложнений. Дополнительно применяют кортикостероиды, которые ослабляют интенсивность пиогенного воспаления — ведущего патогенетического механизма гнойного менингита.
Cерьезным препятствием на пути специфической профилактики H. influenzae-инфекции является слабая иммуногенность b-капсульного материала для детей младшего возраста, которые подвержены максимальному риску заболеть гемофильным менингитом. Проблему удалось решить при помощи конъюгированных вакцин, т.е. b-антигена, ковалентно связанного с белковыми носителями. В современных вакцинах носителями служат дифтерийный или столбнячный анатоксин, а также белки наружной мембраны менингококка группы В. Схемы применения разных вакцин варьируют, но все они предусматривают повторные инъекции, начиная с двухмесячного возраста. Массовая вакцинация, которая проводится во многих странах, резко сократила вероятность местных (эпиглоттит) и системных (менингит) H. influenzae-инвазий.
Детям, находившимся в тесном контакте с больным менингитом, рекомендуется назначение антибиотиков, но этим нельзя злоупотреблять из-за возможности быстрого развития лекарственной устойчивости среди вирулентных штаммов H. influenzae.
Возбудитель коклюша
- Бордетеллы и их лидер.
- Псевдогемофильность и капризы культивирования.
- Адгезины, антифагоцитарные факторы и цитотоксины.
- Патогенетическая многоликость коклюшного токсина.
- Катаральная и пароксизмальная фазы коклюша: противоречия между микробиологией и клиникой.
- Адаптация и болезнетворность B. рertussis.
- Иммунитет и вакцинопрофилактика.
Коклюш — острая высококонтагиозная инфекция респираторного тракта. У непривитых людей, не болевших ранее коклюшем, индекс восприимчивости достигает 0,7—0,75, т.е. из 100 человек, вступивших в тесный контакт с больным, заболевает 70—75. В типичной форме болезнь проявляется приступами (пароксизмами) кашля, тяжесть и длительность которых сильно варьируют. В детском возрасте коклюш протекает как тяжелая, пугающая (нередко терроризирующая) инфекция, которая может закончиться летальным исходом, особенно у детей до 5 лет. Наибольшую опасность коклюш представляет на первом году жизни, когда заболевание осложняется смертельной бронхопневмонией. У взрослых процесс, как правило, ограничивается катаральными симптомами, длительно протекая под маской общей простуды или трахеобронхита.
Коклюш — термин французского происхождения. Согласно одной из версий, он возник из слова «cock» (петух), отражая сходство между звуком при вдохе после завершения приступа кашля (для этого требуется преодолеть сопротивление спазмированных бронхов) и криком петуха. Клинически коклюш принято делить на три периода — катаральный (начальный, продромальный), спазматический (судорожный, пароксизмальный, конвульсивный) и разрешающий (обратное развитие). Иногда дополнительно выделяют период реконвалесценции, который охватывает 2—6 мес после клинического завершения процесса.
Катаральная стадия начинается через 3—14 (обычно 5—8) дней после внедрения возбудителя и протекает как простуда, бронхит или грипп. В этом периоде больной высокозаразен, так как в верхних дыхательных путях присутствует множество коклюшных бактерий в вирулентной фазе (см. ниже). Кашель появляется в конце катарального периода и прогрессирует к концу второй недели спазматической стадии. Приступы кашля иногда настолько сильны, что сопровождаются рвотой и требуют искусственного дыхания. Период выздоровления начинается через 4—8 нед и характеризуется снижением частоты и силы пароксизмов (приступов) кашля. Осложнения включают вторичные бактериальные инфекции респираторного тракта и среднего уха (средний отит), поражение центральной нервной системы (судороги и пирексия, особенно при наслоении вторичных инфекций), паховую грыжу и выпадение прямой кишки (следствие тяжелого кашля). Общая продолжительность болезни 1—3 мес.
Возбудитель коклюша выделен в 1910 г. из мокроты больного ребенка французскими микробиологами Ж.Борде и О.Жангу. По современной классификации, он относится к роду Bordetella (в честь Ж. Борде) и носит название Bordetella pertussis (лат. pertussis — сильный кашель). Кроме B. pertussis род бордетелл включает еще три вида, открытых значительно позднее: B. parapertussis, B. bronchoseptica и B. avium. Подобно классической палочке коклюша, B. parapertussis встречается только у человека, вызывая около 5% всех случаев коклюша, точнее паракоклюша — заболевания, протекающего в атипичной, более мягкой форме. B. bronchoseptica вызывает острые респираторные заболевания у различных животных (кроликов, собак, кошек, свиней, лис, обезьян и др.) и крайне редко (в качестве оппортунистической инфекции) поражает человека. B. avium выделяется от птиц (особенно индюшек), больных ринотрахеитом.
Из сказанного следует, что бордетеллы объединены общим патогенетическим признаком — способностью колонизировать и поражать эпителий респираторного тракта, хотя спектр естественных хозяев у них различен. Гомология ДНК бордетелл настолько высока (более 90%), что их вполне можно было бы объединить в один вид. Но это тот случай, когда малозаметная генетическая дивергенция определила фенотипическое своеобразие бактерий, которое имеет принципиальное экологическое и патогенетическое значение. В медицинской микробиологии приоритетными для изучения являются бактерии Борде—Жангу, т.е. B. pertussis.
Микробиологический профиль
B. pertussis относится к весьма деликатным бактериям. Это мелкие грамотрицательные палочки, которые не образуют спор, неподвижны, в S-форме имеют нежную капсулу. Они не устойчивы во внешней среде, а прогревание при 55°С убивает их через 30 мин. Единственный природный резервуар инфекции — человек, и лишь он болеет коклюшем. В эксперименте, инокулируя взвесь живых B. pertussis в трахею, можно вызвать катаральное воспаление респираторного тракта у животных (щенят, котят, обезьян и др.), но типичной картины коклюша с пароксизмальным кашлем воспроизвести не удается.
Для выделения B. pertussis требуются специальные среды. История их создания заложена пионерскими работами Борде и Жангу, которые использовали картофельно-глицерино-кровяной агар. Добавление 15—20% крови было обязательным, на основании чего возбудитель коклюша долгое время числился среди гемофилов, для размножения которых требуются два ростовых фактора — гемин и НАД (см. «Палочка инфлюэнцы»). Позже оказалось, что причиной «гемофильности» бордетелл служит не их потребность в ростовых факторах, а высокая чувствительность к ненасыщенным жирным кислотам, сульфидам и перекисям, которые накапливаются в процессе роста бактерий и оказывают на них токсическое действие. Кровь выполняет роль сорбента для этих метаболитов, но вместо нее можно использовать ионообменные смолы или активированный уголь. Кстати, последний в составе различных питательных сред (например, казеиново-угольный агар) широко применяется для культивирования бордетелл. Даже в оптимальных условиях B. pertussis растет медленно, и лишь спустя 2—3 дня удается обнаружить мелкие колонии, окруженные небольшой зоной гемолиза. Будучи аэробом (это способствует выживанию в обильно аэрируемом респираторном тракте), коклюшная палочка лучше растет в атмосфере, обогащенной углекислым газом (5—7% СО2), т.е. отличается капнофильностью.
Факторы патогенности
Коклюшная палочка обладает сложным набором факторов патогенности, действующих согласованно и целенаправленно. Основной точкой их приложения является мерцательный эпителий респираторного тракта. B. pertussis практически лишен инвазивности. Попав в дыхательные пути, возбудитель прикрепляется к ресничкам мерцательного эпителия трахеи и бронхов и размножается на их поверхности, вызывая повреждение эпителиоцитов (рис. 1, 2).

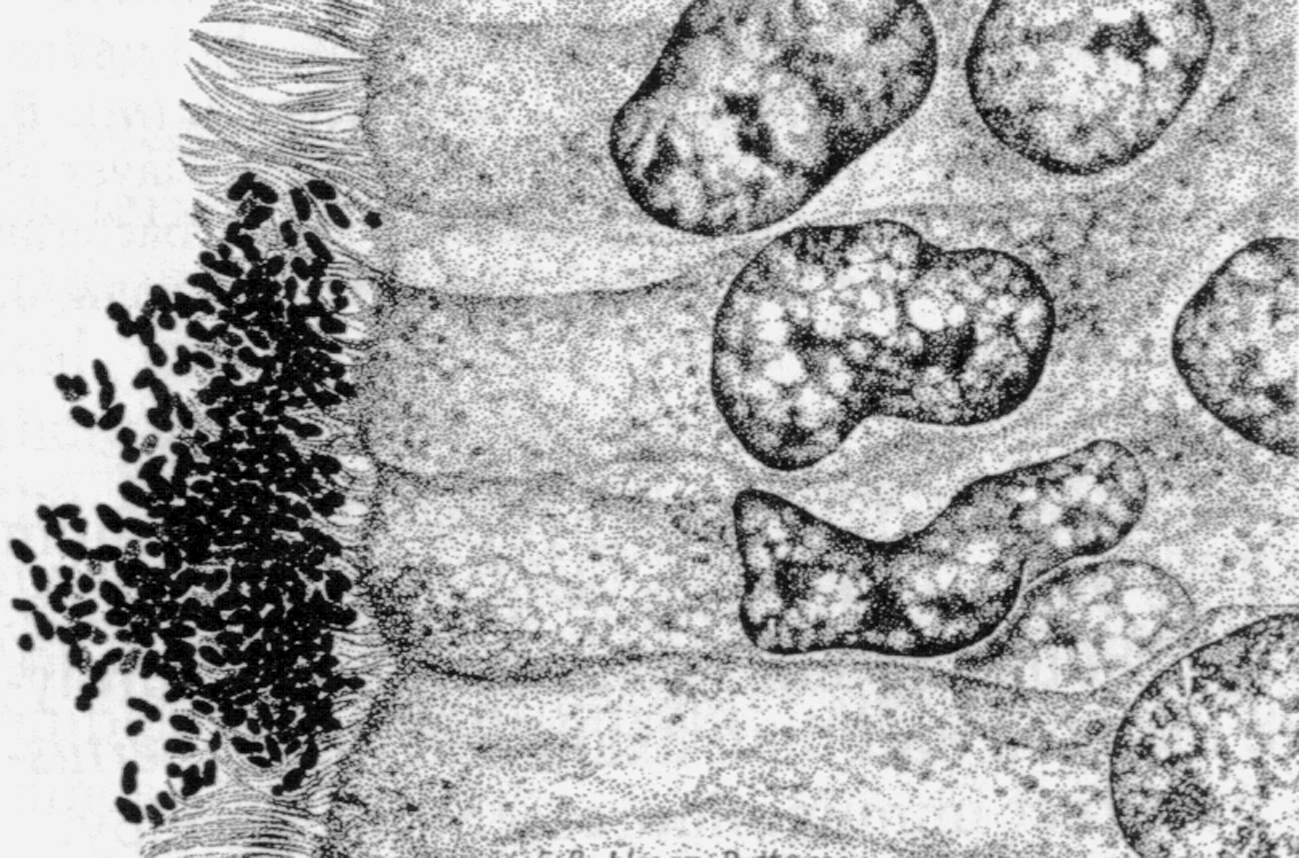

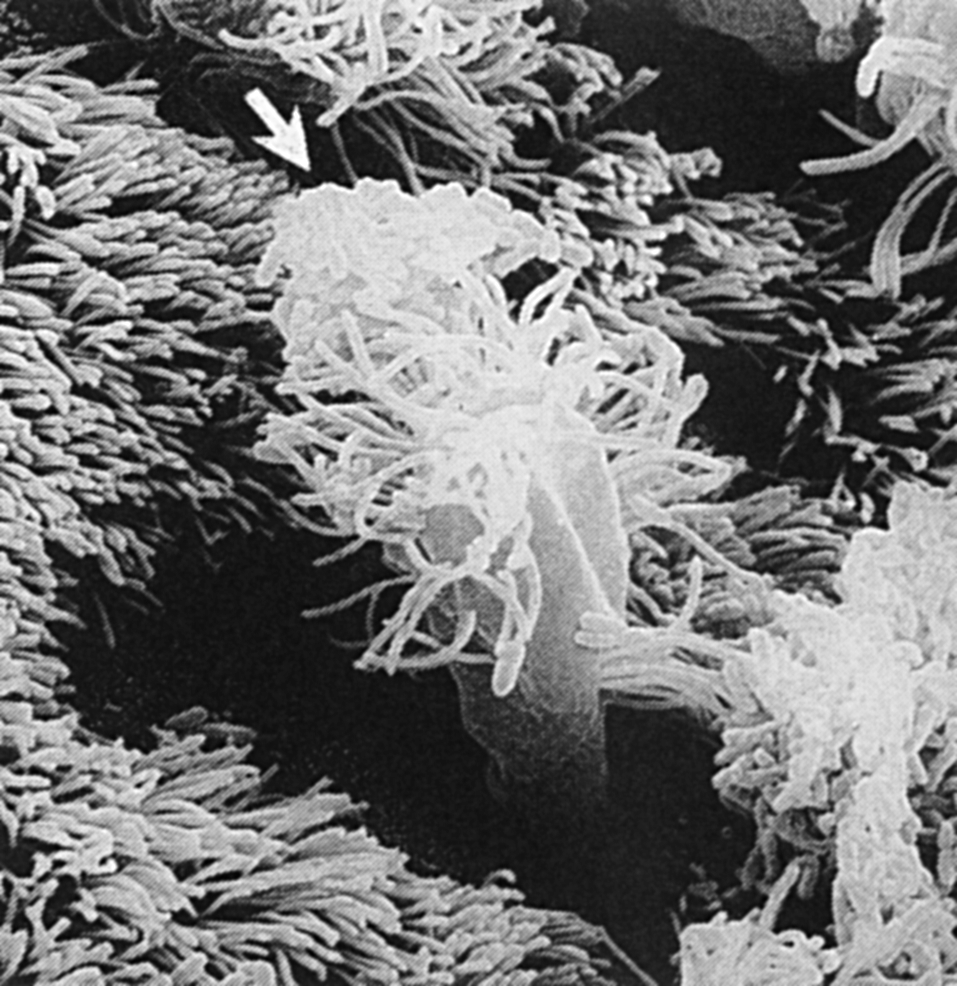
Повреждение не бывает значительным, инфекция никогда не проникает в глубоколежащие ткани, а тем более в кровяное русло. Это означает, что в патогенезе коклюшной инфекции большую роль играют функциональные извращения в клеточных системах респираторного тракта. Они связаны со специфическими токсинами возбудителя и сохраняются после его удаления из организма. В этом парадокс коклюшной инфекции: клинические симптомы усиливаются на фоне снижения концентрации возбудителя в зоне поражения. Подобно многим грамотрицательным бактериям, B. pertussis содержит гены, кодирующие аппарат секреции III типа, однако они не включаются в работу. Этим коклюшная палочка отличается от своего «прародителя», B. bronchоseptica. Причины такой эволюции (точнее инволюции) остаются неясными.
Адгезия. Базисным патогенетическим механизмом при коклюше является адгезия возбудителя на мерцательном эпителии респираторного тракта. Коклюшная палочка располагает комплексом адгезинов, основное значение среди которых имеют филаментозный гемагглютинин (ФГ), белки наружной мембраны (пертактин, ВrkA) и фимбрии (пили). Приоритет отдается ФГ, остальные адгезины играют вспомогательную роль. Не случайно ФГ используется в качестве одного из компонентов бесклеточной противококлюшной вакцины.
ФГ представляет собой белок, агглютинирующий эритроциты и структурно оформленный в виде поверхностных микрофиламентов. Он содержит трипептид — аргинин-глицин-аспарагиновую кислоту (RGD), который определяет связывание с интегринами, углеводными и сульфатидными группировками на поверхности реснитчатых клеток, а также макрофагов и нейтрофилов (такой же активный центр имеют адгезивные белки наружной мембраны — пертактин и BrkA). В экспериментах на животных введение в трахею аналогов лактозы предотвращает колонизацию респираторного тракта коклюшными бактериями. Аналогичный эффект вызывают анти-ФГ-антитела и антитела против клеточных мембранных лактозилцерамидов.
Токсины. Закрепление в респираторном тракте, за которым следует размножение бактерий, создает условия для реализации токсического потенциала коклюшной палочки. Он складывается из комплекса факторов, оказывающих местное и системное воздействие на организм. Главной мишенью остается мерцательный эпителий респираторного тракта.
Трахеальный цитотоксин. Это фрагмент пептидогликана, который является промежуточным элементом его синтеза или продуктом деградации (рис. 3). Каким образом, не обладая прямой цитотоксичностью, пептидогликан повреждает эпителиоциты и почему именно коклюшная палочка, которая по структуре пептидогликана принципиально не отличается от других грамотрицательных бактерий, обладает этим механизмом деструкции? Одной из причин является то, что пептидогликан и его производные побуждают макрофаги и эпителиоциты слизистой оболочки к секреции цитокинов и других медиаторов, которые потенциально опасны для реснитчатого эпителия. В отличие от большинства бактерий, прочно удерживающих пептидогликан, коклюшная палочка постоянно высвобождает его субкомпоненты, которые включаются в опосредованную цитотоксичность.

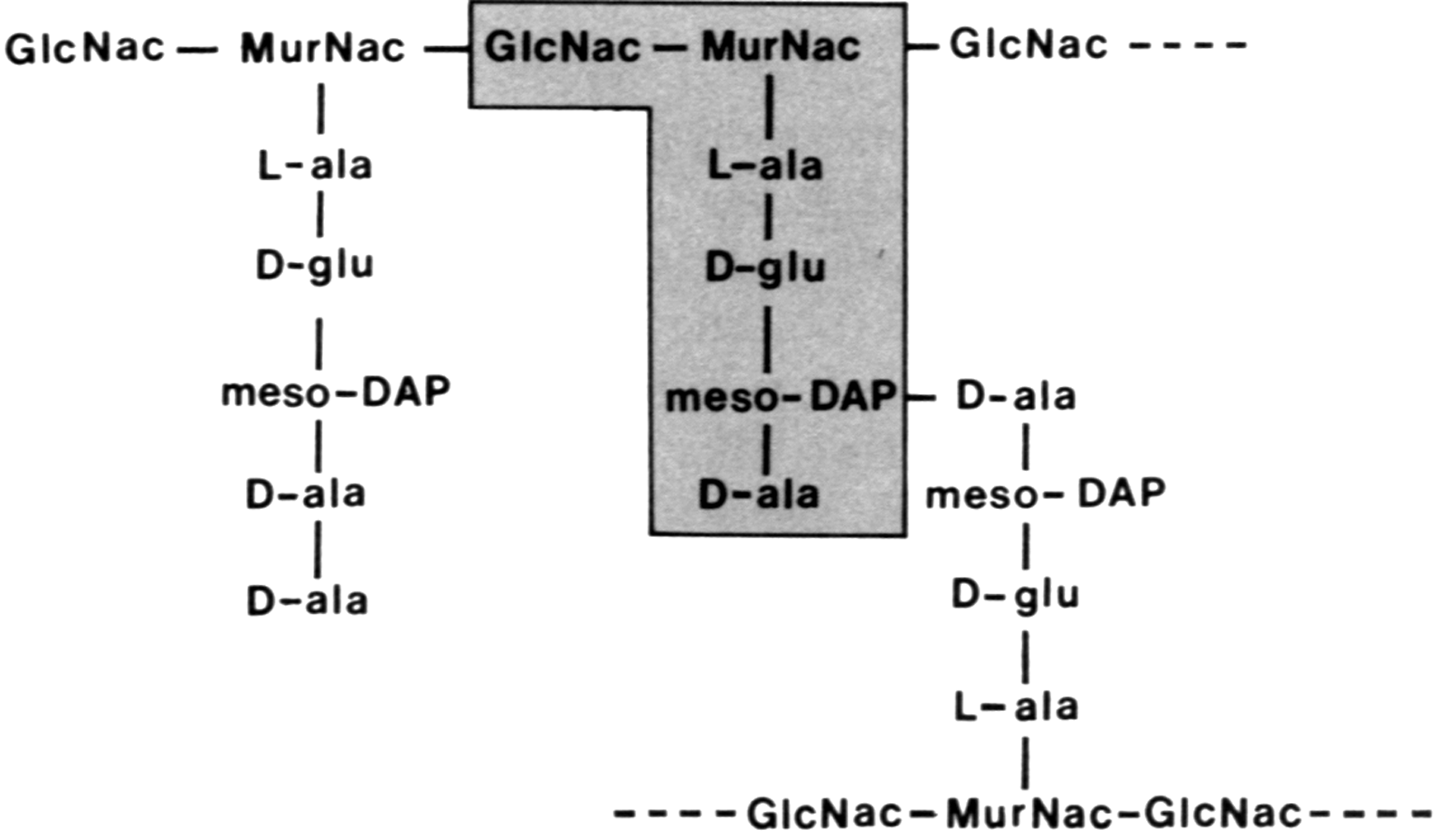
Дермонекротоксин (известен еще как термолабильный токсин — инактивируется при 56°С) индуцирует некроз при внутрикожном введении, вызывая ишемию, обусловленную спазмом артериол. Инициируя повреждение, участвует в возбуждении воспалительной реакции, которая лежит в основе первой (катаральной) фазы болезни.
Коклюшный токсин (термостабильный токсин — инактивация при 80°С). Несмотря на наличие других токсических продуктов (остальные бордетеллы лишены этого признака), именно его называют коклюшным токсином, Ptx (син.: лимфоцитоз-стимулирующий фактор, пертуссиген, фактор активации островковой функции поджелудочной железы, гистаминсенсибилизирующий фактор). Ptx секретируется в два этапа. Сначала происходит транспозиция его субкомпонентов в периплазматическое пространство, откуда после сборки токсин выделяется при помощи системы секреции, близкой секреции IV типа.
Ptx действует не только как токсин, но и как фактор адгезии, cвязывающийся с мерцательным эпителием респираторного тракта. Полагают, что это главный индуктор пароксизмальной фазы заболевания. Вместе с тем, до сих пор не найдено экспериментальной модели, на которой бы удалось воспроизвести пароксизмальный кашель. Однако ряд других патологических сдвигов, характерных для коклюша (повышенная чувствительность к гистамину, лимфоцитоз, гипогликемия), можно вызвать у животных при помощи очищенных препаратов коклюшного токсина. Обезвреженный Ptx (анатоксин) входит в состав бесклеточной вакцины, используемой для профилактики коклюша.
Концепция о полифункциональном токсине, определяющем клиническую специфику коклюша, базируется на патогенетической универсальности молекулярной мишени для коклюшного токсина в поражаемых клетках. Подобно ряду других бактериальных токсинов коклюшный токсин принадлежит к А/В категории АДФ-рибозилтрансфераз — ферментов, которые после проникновения в клетку отщепляют никотинамид от НАД, присоединяя остаток аденозиндифосфорибозы к белкам-мишеням (см. «Возбудитель дифтерии»). По аналогии с холерным энтеротоксином (холерогеном), АДФ-трансферазная активность каталитического фрагмента коклюшного токсина нацелена на семейство так называемых G-протеинов — ферментов, занимающих ключевую позицию в реализации рецепторзависимых сигналов. АДФ-рибозилирование G-белков (это связано с А-субкомпонентом токсина) выводит из-под негативного контроля аденилатциклазу, вызывая повышение внутриклеточной концентрации циклического АМФ и извращение многих клеточных функций. Избирательность клеточных мишеней зависит от сродства к определенным мембранным структурам: подобно филаментозному гемагглютинину, коклюшный токсин наиболее активно рецептируется лактозилцерамидами реснитчатого эпителия респираторного тракта.
Впрочем, уверенность в болезнетворном значении коклюшного токсина не снимает ряд неясностей относительно уникального эффекта коклюшной палочки, в частности эволюции пароксизмального кашля. Более того, есть факты, которые заставляют думать, что инфекция оставляет такие изменения в слизистой оболочке бронхиального дерева, которые повышают реактивность к раздражителям, безвредным для нормального респираторного тракта. Возможно, одной из причин служит снижение порога чувствительности к гистамину, хотя (повторим!) смоделировать коклюшный кашель на животных не удается, несмотря на воспроизведение других симптомов коклюшной интоксикации, включая гиперреактивность к гистамину. Cуществует даже мнение о том, что Ptx не участвует в формировании пароксизмального кашля при коклюше. По крайней мере, B. Parapertussis, несмотря на отсутствие Ptx-продукции, вызывает такой же кашель, какой наблюдается при настоящем коклюше.
Аденилатциклаза. Была открыта как фактор с гемолитическими свойствами. Позже оказалось, что это токсин, который, действуя контактно на различные клетки (большая часть аденилатциклазы остается на бактериях в связанном состоянии), переводит АТФ в циклический АМФ. Это, в частности, выключает многие функции фагоцитарных клеток (хемотаксис, поглощение, продукцию активированного кислорода), необходимые для уничтожения возбудителя. Интересно, что активация коклюшной аденилатциклазы происходит только в присутствии кальмодулина — кальцийсвязывающего белка эукариотических клеток. Такой пример уникален для B. pertussis.
Липополисахарид. Необычность приступов коклюшного кашля побуждала в прошлом обращаться к идеям нервизма, связывая коклюш с патологией центральной нервной системы. Даже в современной литературе встречаются довольно расплывчатые упоминания о кашлевых рецепторах, которые неким образом сенсибилизируются метаболитами коклюшной палочки. К сожалению, сегодня трудно сказать нечто большее об этом загадочном явлении, за которым скрываются уникальные свойства возбудителя, диктующие своеобразие его взаимоотношений с хозяином. В любом случае на коклюш нельзя смотреть как на моноинтоксикацию. Токсический эффект может быть результатом суммарной активности нескольких факторов, действующих на местном и системном уровнях. Не последнюю роль в этом ансамбле играет липополисахаридный эндотоксин, который (как и у всех грамотрицательных бактерий) входит в состав наружной мембраны коклюшной палочки и несет антигенные эпитопы, общие для всех бордетелл (родоспецифический антиген).
Изменчивость
Характерной особенностью бордетелл является фенотипическая изменчивость. Принципиально, что она распространяется на признаки, связанные с вирулентностью. Это наблюдается в культурах in vitro и в инфицированном организме, отражая реакцию бактерий на сигналы из внешней среды. Само понятие «фазовые вариации», хорошо известное микробиологам, введено в 1931 г. для обозначения культуральных и антигенных модуляций B. pertussis.
Изменение культуральных свойств коклюшной палочки предложено характеризовать четырьмя фазами. Переход от фазы I к фазам II—IV соответствует S—R-трансформации, в ходе которой бактерии претерпевают ряд структурно-функциональных изменений, а главное — становятся авирулентными.
Вирулентные штаммы (S-варианты) изолируются обычно в раннем (катаральном) периоде заболевания. Они продуцируют токсины, располагают факторами, необходимыми для колонизации респираторного эпителия, имеют видоспецифические и общие для бордетелл наружные антигены. Их наличие определяет патогенетический потенциал коклюшных бактерий и высокую способность инфицировать других людей. Экспрессия генов, ответственных за образование факторов вирулентности, подчинена стратегии оптимальной адаптации бактерий к внешней среде и в конечном счете нацелена на выживание.
У коклюшной палочки отлично работает механизм, получивший название глобальной регуляции генов вирулентности. Переключение с вирулентного на невирулентный фенотип находится под контролем центрального регуляторного гена, активность которого меняется в зависимости от среды, где оказываются бактерии. Вирулентные штаммы быстро становятся авирулентными (R-формы), если их культивировать при 25°С, т.е. в режиме, близком к температуре носовых ходов. Возврат вирулентности (переход в фазу I) происходит при 37°С, т.е. при внутренней температуре тела. Это означает, что бактерии коклюша чувствуют снижение температуры, останавливая работу более 20 генов, детерминирующих вирулентный фенотип. Это объясняется тем, что, подобно другим бактериям, возбудитель коклюша имеет механизм передачи сигналов, конвертирующих внешние раздражители в экспрессию определенных генов. Он включает сенсорные белки плазматической мембраны, которые при стимуляции активируют внутриклеточные медиаторы, растормаживающие гены вирулентности. Для коклюшной палочки главным стимулом служит повышение температуры окружающей среды. В авирулентном состоянии она способна персистировать в носовых ходах, так как не продуцируя токсинов и антигенов, ассоциированных с болезнетворностью, не вызывает сильной реакции хозяина и не атакуется накопившимися к данному моменту антителами. Это способствует длительному носительству и распространению возбудителя. Есть данные, что коклюшная палочка может персистировать в макрофагах респираторного тракта, куда она проникает благодаря связыванию филаментозного гемагглютинина макрофагальными рецепторами (взаимодействие между RGD гемагглютинина и СD11b/CD18-интегринами плазматической мембраны макрофагов).
Иммунитет и специфическая профилактика
Перенесенная инфекция оставляет достаточно прочный иммунитет, который предохраняет от рецидивов типичного (пароксизмального) коклюша, но спустя несколько лет не исключает повторения инфекции в катаральной форме. Такие больные представляют наибольшую эпидемиологическую опасность, так как заболевание не диагностируется и противоэпидемические мероприятия не проводятся. В крови больных определяются агглютинирующие, преципитирующие, бактерицидные и комплементсвязывающие антитела, но они появляются поздно (через 3—4 нед после начала заболевания), а потому имеют ограниченное диагностическое значение. Сформулируем и парадокс бактериологической диагностики коклюша: возбудитель нередко исчезает из респираторного тракта раньше, чем кончается болезнь. Более того, вероятность его выделения в классическом (пароксизмальном) периоде гораздо ниже, чем в катаральной фазе заболевания. О причинах (они связаны с патогенетическим своеобразием коклюшной инфекции) говорилось выше, и мы вновь возвращаемся к этому, лишь вспоминая вопросы бактериологов-практиков, потерявших уверенность в надежности бактериологического подтверждения коклюша.
Исходя из представлений о патогенезе коклюша эффективная иммунная защита против его возбудителя должна включать два принципиальных механизма: препятствовать закреплению бактерий на мерцательном эпителии респираторного тракта (антиадгезивное действие) и обеспечивать нейтрализацию токсинов. До сих пор для иммунопрофилактики коклюша используют цельные убитые нагреванием бактерии (в фазе I), обычно в сочетании с дифтерийным и столбнячным анатоксинами, адсорбированными на гидроокиси алюминия (АКДС-вакцина). Широкая вакцинация детей, начавшаяся в 1940-х гг., позволила значительно снизить заболеваемость и смертность от коклюшной инфекции. Однако ряд побочных (поствакцинальных) реакций делает препарат далеко не идеальным. Значительная прослойка детей по разным причинам остается невакцинированной, рискуя заразиться коклюшем и заразить других. Именно это позволяет считать коклюш одной из самых опасных инфекций детского возраста. Расшифровка патогенетически значимых факторов коклюшной палочки даст возможность создания более совершенных препаратов, нацеленных на избирательную индукцию антиадгезивного и антитоксического иммунитета. Один из них уже широко используется за рубежом. Речь идет о бесклеточном препарате, приготовленном на основе коклюшного филаментозного гемагглютинина и детоксицированного коклюшного токсина (Ptx). В США такая вакцина (она получила название аР, от англ. acellular pertussis) уже вытеснила препарат из цельных клеток.
Коклюшная палочка сохраняет чувствительность ко многим антибиотикам. С этой точки зрения этиотропное лечение коклюша не должно было бы иметь принципиальных сложностей. Однако в действительности эффективность антибиотикотерапии оставляет желать лучшего. Антибиотики, убивающие B. pertussis in vitro, не оказывают радикального влияния на пароксизмальную стадию, хотя снижают тяжесть и длительность заболевания. Это согласуется с представлениями о приступах кашля как о следовой реакции, своего рода последействии коклюшных токсинов на фоне снижения концентрации и вирулентности возбудителя в респираторном тракте. По-видимому, по той же причине не эффективен и противококлюшный иммуноглобулин, на который возлагались большие надежды.
Синегнойная палочка
- Псевдомонады.
- Убиквитарность и устойчивость во внешней среде.
- Экологические и эпидемиологические аспекты синегнойной инфекции.
- Пассивная и агрессивная инвазивность.
- Токсины.
- Инвазии и интоксикации.
- Иммунитет и проблемы этиотропной терапии.
Окрашивание гноя в сине-зеленый цвет (особенно часто это наблюдали в ожоговых стационарах) интриговало задолго до утверждения в медицине микробиологической науки. Предположение о микробной природе этого явления высказано Дюке в 1862 г., а чистая культура синегнойной палочки выделена Гессардом в 1882 г.
Долгое время на синегнойную палочку смотрели как на безвредный сапрофит, который, контаминируя раны, лишен патогенетических потенций. Пренебрежительному отношению содействовало резкое сокращение синегнойной инфекции после введения антисептики и асептики. Вместе с тем, уже в начале ХХ столетия в руководствах по микробиологии указывалось, что широкое распространение синегнойной палочки в окружающей среде, на коже, слизистых оболочках и в пищеварительном тракте человека дает основания опасаться «завоевания синегнойной палочкой хирургических отделений, где она иногда держится с таким упорством, что требуется тщательная дезинфекция с эвакуацией больных». Это предостережение сбылось в наши дни, когда широкое использование инвазивных диагностических и лечебных процедур в сочетании с необыкновенной устойчивостью синегнойной палочки к антибактериальным препаратам и антисептикам, убиквитарностью и удивительной непритязательностью к питанию подняли риск контаминации и вероятность клинически значимой инфекции.
«Синегнойка» (так часто именуют синегнойную палочку в больничном обиходе) стала причиной одной из устрашающих госпитальных инфекций в стационарах хирургического профиля. Как правило, это вторичная пиогенная инфекция, осложняющая основное заболевание. Иными словами, синегнойная палочка является типичным представителем сапрофитических микробов-оппортунистов, о патогенетической тактике которых уже не раз говорилось выше.
Синегнойная палочка принадлежит к одному из самых обширных семейств бактерий — псевдомонадам. Это грамотрицательные, подвижные палочки, не образующие спор. Характерная особенность — полярное расположение одного или двух жгутиков. Благодаря им синегнойная палочка выглядит как высокоподвижный микроб, с удивительной скоростью распространяющийся в поле зрения микроскопа (рис. 1). В этом отношении псевдомонады напоминают бактерии семейства Vibrionaceae, в частности холерный вибрион. Если не считать данного признака, то по морфологии псевдомонады похожи на энтеробактерии.

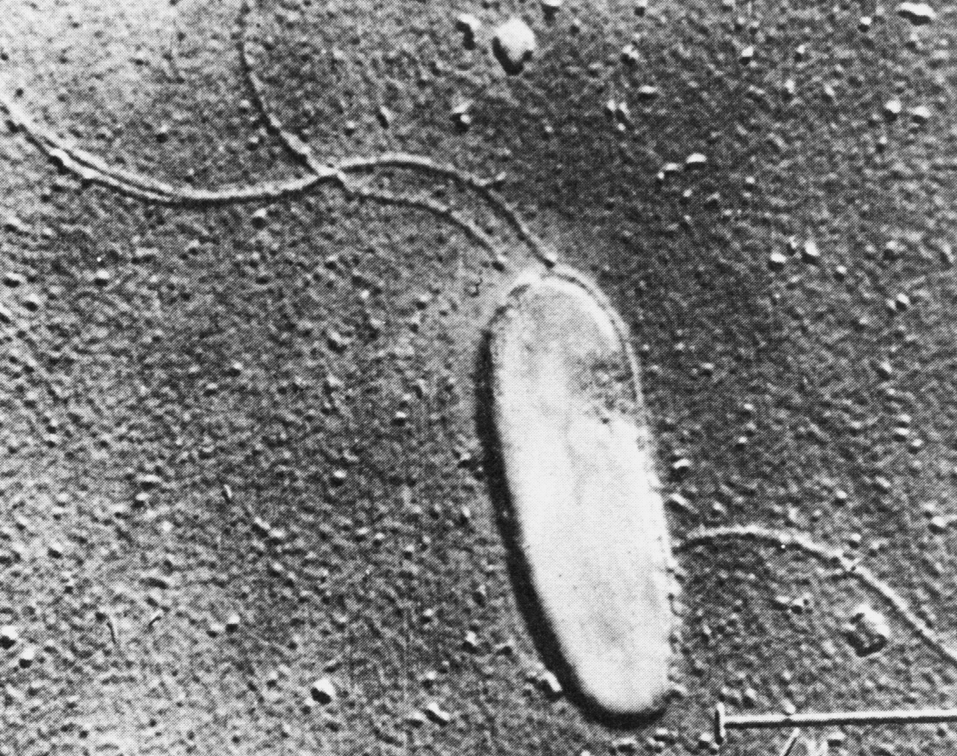
Наименование семейства (Pseudomonadaceae) происходит от двух греческих слов: pseudes — ложный, сходный и monas — название группы простейших с полярными жгутиками (monad — неделимое целое; одноклеточный организм). Семейство включает несколько родов и огромное число видов, которые в своем большинстве являются аэробными сапрофитами, свободно живущими в воде и почве. Некоторые псевдомонады паразитируют на растениях, обладая фитопатогенностью. Псевдомонады называют «неферментирующими» бактериями, подчеркивая тем самым, что это строгие аэробы с респираторным типом энергетического метаболизма, для которых молекулярный кислород служит необходимым акцептором электронов на завершающей стадии окислительно-восстановительных реакций, генерирующих АТФ. Впрочем, большинство штаммов могут расти в бескислородных условиях, используя нитрат в качестве конечного акцептора электронов. Благодаря поразительной всеядности (в качестве источника углерода и энергии ими могут использоваться почти все природные органические соединения) псевдомонады являются убиквитарными мусорщиками, ответственными за аэробную минерализацию самого разнообразного органического материала. Фитопатогенные разновидности (главным образом ксантомонады) наносят большой ущерб сельскому хозяйству, деревообрабатывающей и другим видам промышленности, где используется растительное сырье. Псевдомонады служат одной из самых распространенных причин порчи продуктов (яйца, вареное мясо, рыба, молоко), тем более что многие из них (включая синегнойную палочку) способны расти при низких температурах.
Синегнойная палочка относится к роду Pseudomonas, который включает более 150 видов бактерий. Лишь немногие из них патогенны для человека. Наряду с безусловным лидером, синегнойной палочкой, причиной пиогенных осложнений могут быть Ps. putida, Ps. fluorescens, Ps. puckettii, а также Ps. cepacia, недавно переименованный в Burkholderia cepacia. Кроме того, опасность представляют Ps. mallei и Ps. pseudomallei — возбудители сапа и миелоидоза (теперь они называются Burkholderia mallei и Burkholderia pseudomallei). Из них лишь возбудитель сапа — облигатный паразит животных, остальные (подобно большинству псевдомонад) — свободноживущие сапрофиты, для которых паразитизм необязательное, случайное явление.
Синегнойная палочка принадлежит к немногим бактериям, которые в естественных условиях патогенны не только для человека, но и для животных. Она вызывает у них гнойные очаговые процессы, поражения кишечника и даже сепсис.
Микробиологический профиль
Научное название синегнойной палочки — Pseudomonas aeruginosa. Видовой эпитет отражает цвет колоний на питательных средах (лат. aeruginosa — оттенок медной ржавчины). Впрочем, для медицинской микробиологии термин Bacterium pyocyaneum (греч. pyos — гной, cyaneum — синий), выглядит более привычным, а главное — патогенетически осмысленным.
Синегнойная палочка обладает двумя характерными признаками, которые неизбежно обращают на себя внимание при культивировании бактерий и были известны задолго до открытия возбудителя, так как наблюдаются в клинике при синегнойной инфекции. Речь идет об образовании водорастворимых сине-зеленых пигментов, диффундирующих в окружающую среду (агар вокруг колоний, перевязочный материал), и о своеобразном запахе, сопровождающем рост бактерий. Он напоминает запах цветущей липы, хотя и в шаржированном виде.
Из набора пигментов, которыми богаты псевдомонады, специфичным для синегнойной палочки является пиоцианин — сине-зеленый феназиновый пигмент. Кроме того, синегнойная палочка продуцирует желто-зеленый флюоресцеин (пиовердин), общий для группы флюоресцирующих (в ультрафиолете) псевдомонад, и менее типичный пиорубин — пигмент с красным оттенком. Интенсивность пигментообразования зависит от условий, но встречаются штаммы, лишенные этого признака. В таких случаях надежным диагностическим критерием служит проба на оксидазу с материалом, взятым непосредственно из колоний. Она положительна для большинства псевдомонад, отличая их от сходных бактерий, прежде всего энтеробактерий.
Еще один существенный культуральный признак — образование слизи, делающей колонии мукоидными (слизистыми), а бульонные культуры — вязкими. Такие штаммы обычно выделяются из мокроты и редко — из очагов раневой инфекции. Скорее всего речь идет о селекции мутантов, имеющих повреждение одного из генов (mucA), ингибирующих продукцию слизи, хотя не исключены и другие механизмы, связанные, в частности, с фазовыми вариациями alg-генов, детерминирующих продукцию мукоидного материала (алгината). Слизь по своим химическим (полисахарид типа алгината) и функциональным (антифагоцитарная активность) свойствам близка капсуле, но в отличие от нее не является структурным компонентом клетки, а диффундирует в окружающую среду (экзополисахарид).
Подобно всем бактериям, у синегнойной палочки есть бактериоцины, называемые пиоцинами, а также бактериофаги. И те и другие используются в качестве эпидемиологических маркеров при изучении закономерностей распространения внутрибольничной синегнойной инфекции.
Много работ посвящено изучению антигенной структуры синегнойной палочки. Они были нацелены на серотипирование штаммов и поиск протективных антигенов, пригодных для иммунопрофилактики и иммунотерапии синегнойной инфекции. Диагностически значимыми являются О- и Н-антигены, т.е. компоненты липополисахаридного комплекса (эндотоксина) наружной мембраны и жгутиков. Наиболее полная схема серотипирования включает около 30 О-антигенов и 10 Н-антигенов, сочетанный анализ которых позволяет дифференцировать более 50 сероваров, используя их в качестве эпидмаркеров. Известны и другие антигены синегнойной палочки, антитела к которым легко получить при иммунизации животных и выявить у больных синегнойной инфекцией. Особенно впечатляет спектр антител при хроническом поражении легких у больных муковисцидозом: нередко он представлен антителами более чем к 60 антигенам синегнойной палочки.
Экологические и эпидемиологические аспекты
Клинико-эпидемиологическое значение синегнойной палочки определяется суммой ее экологических и патогенетических признаков — убиквитарностью и неприхотливостью, устойчивостью к антибактериальным дезинфектантам и лечебным препаратам, пиогенностью и продукцией деструктивно-токсических начал. Вместе с тем, как уже говорилось, синегнойная палочка — типичный микроб-оппортунист, болезнетворность которого реализуется лишь при столкновении с ослабленным организмом, неспособным оказать достойное сопротивление возбудителю. Синегнойная палочка может колонизировать различные участки тела, но для инвазии и стабилизации очага инфекции требуются повреждение кожи и слизистых оболочек, снижение их колонизационной резистентности (например, на фоне трофических расстройств), заражение большими дозировками (инвазивные контакты с инфицированными инструментами). Этим объясняются частое инфицирование ожоговых и хирургических ран, пролежней, трофических язв; развитие постинъекционных абсцессов, септических осложнений после внутрисосудистых манипуляций; поражение мочевыводящей системы при пользовании нестерильными катетерами; пневмонии у больных с искусственной вентиляцией легких и пр. Повышенному риску подвержены больные, в лечении которых вынужденно применяется антиметаболитная и иммуносупрессивная терапия (онкологический контингент, реципиенты аллогенных трансплантатов), новорожденные и дети младшего возраста, старики.
Устойчивость и непритязательность к питанию определяют почти универсальное распространение синегнойной палочки в больничной среде, создавая широкие возможности для формирования госпитальных штаммов. Практически единственным условием для ее выживания является достаточная влажность. Синегнойную палочку выделяют из раковин, кранов, посуды, пищи, респираторов, систем кондиционирования воздуха, аппаратов искусственного кровообращения и другого медицинского инструментария, препаратов крови и кровезаменителей. Имеет значение и ее способность размножаться в широком диапазоне температур, причем некоторые штаммы растут даже при 4°С, создавая опасность контаминации препаратов, хранящихся в холодильнике. Источником, который легко просмотреть, служит дистиллированная вода: в ней синегнойная палочка не только выживает, но и размножается за счет минимальных загрязнений. Описаны случаи заражения от комнатных растений, цветов, что вполне объяснимо, так как микроб постоянно присутствует в почве и обладает потенциальной фитопатогенностью.
Устойчивость к дезинфектантам позволяет синегнойной палочке не только выживать, но и размножаться в разбавленных растворах антисептиков (например, в фурацилине, риваноле, детергентах). Она малочувствительна к оксидантам, в том числе к активным формам кислорода, выживая в системах, генерирующих синглетный кислород, где другие бактерии быстро погибают (например, в средах, где происходят реакции с фотодинамическим эффектом). Здесь сказываются особенности, общие для псевдомонад — облигатная аэробность с мощной антиоксидантной защитой и наличие окислительных ферментов (монооксидаз), модифицирующих самые неудобоваримые субстраты.
Эпидемически значимым резервуаром внутрибольничной синегнойной инфекции являются медицинский, обслуживающий персонал и сами больные. Синегнойная палочка присутствует в кишечнике примерно 10% здоровых людей и спорадически попадает на кожу. Уровень инфицированности возрастает, начиная с первых дней пребывания больных в стационаре. К 3-му дню он иногда достигает 30%, а в дальнейшем повышается до 40%. Это означает, что синегнойная инфекция обычно возникает в результате экзогенного заражения, хотя нельзя недооценивать и роль аутоштаммов.
Резистентность к антибиотикам
Общеизвестна высокая лекарственная резистентность синегнойной палочки. Как и у других бактерий, она связана с конституциональной нечувствительностью к ряду антибактериальных препаратов и с адаптивной (индуцибельной) устойчивостью, приобретаемой благодаря R-плазмидам. Первый вариант определяется особенностями внешних структур синегнойной палочки (блокада внутриклеточной диффузии антибиотиков отрицательно заряженным материалом, обволакивающим бактерии), а также ферментативной модификацией антибактериальных агентов. Имеет значение построение биопленочных образований, особенно при инфицировании муковисцидозных легких.
Что касается адаптивной резистентности, то у синегнойной палочки имеется набор плазмид, детерминирующих невосприимчивость к бета-лактамным антибиотикам, аминогликозидам, хлорамфениколу, сульфаниламидам и некоторым антисептикам. Часть таких плазмид (точнее, компонующих их R-транспозонов) заимствована у других бактерий, прежде всего у энтеробактерий. Вместе с тем, число плазмидосодержащих штаммов среди клинических изолятов синегнойной палочки невысоко — около 20%, хотя по сравнению с другими бактериями, располагающими сходными R-плазмидами, синегнойная палочка гораздо устойчивее. Это означает, что ее резистентность — прежде всего конституциональный признак, детерминируемый хромосомными генами на уровне вида и даже рода псевдомонад. Показателен пример с имипенемом, новым бета-лактамным антибиотиком, который проникает в периплазматическое пространство синегнойной палочки через специфический порин — OprD. Резистентность к имипенему развивается благодаря мутациям, приводящим к потере OprD; бета-лактамазная устойчивость не имеет решающего значения.
Подавление сопутствующей микрофлоры при длительном применении антибиотиков ведет порой к тому, что синегнойная палочка остается единственным видом бактерий в очаге инфекции, препятствуя заживлению ран. Впрочем, конкурентоспособность псевдомонад может включать и элементы агрессивности. Давно замечена способность фильтрата многосуточных бульонных культур синегнойной палочки лизировать различные бактерии — дифтерийную палочку, холерный вибрион, стафилококки, стрептококки, бациллы сибирской язвы, возбудителей брюшного тифа, чумы. В конце ХIХ — начале ХХ вв. частично очищенный фильтрат синегнойной палочки (пиоцианаза) даже пробовали использовать в лечении инфекционных заболеваний. Попытки парентерального введения были быстро оставлены из-за токсичности препарата; более успешно пиоцианаза применялась как наружное средство для лечения дифтерии и гнойно-воспалительных процессов. Кроме бактерицидного начала, препарат, по-видимому, действовал за счет протеолитических ферментов, которыми богата синегнойная палочка (см. ниже). Этим, в частности, можно объяснить эффективность пиоцианазы в удалении дифтеритических пленок при дифтерии. В конечном счете препарат был оставлен, разделив судьбу многих антибактериальных веществ, активных in vitro, но не пригодных для лечебных целей. Так или иначе, но это был один из первых опытов клинической реализации идеи об антибиозе. Позже было установлено, что одним из бактерицидных начал пиоцианазы является пиоцианин, хотя суммарный эффект не исчерпывается пигментами феназинового ряда.
Факторы патогенности
Патогенность синегнойной палочки определяется ее способностью 1) инвазировать и разрушать ткани; 2) персистировать в них, поддерживая воспаление; 3) вызывать общую интоксикацию организма. Это пиогенные бактерии, особенностью которых является мощная гидролазная активность, нацеленная на клеточные мембраны и матриксные белки соединительной ткани, в сочетании с системным токсическим эффектом.
Подобно всем пиогенным бактериям, синегнойная палочка является внеклеточным патогеном, вызывая реакцию нейтрофилов и одновременно оказывая им сопротивление. От этого зависит весь ход событий, начало и итог синегнойной инфекции. Антифагоцитарными (антинейтрофильными) свойствами обладают капсулоподобный экзополисахарид, пили, липополисахарид наружной мембраны, фосфолипазы. Три главные протеазы синегнойной палочки (две эластазы и щелочная протеаза) вкупе обладают широким спектром протеолитической активности. Они разрушают эластин, коллаген, ламинин, иммуноглобулины, факторы комплемента (C1q, C3, С5), лизоцим секретов, белки роговицы, гамма-интерферон. Перевес бактерий (и соответственно неэффективность нейтрофилзависимого воспаления) ведет к стабилизации инфекции, усилению инвазивного процесса и интоксикации. Расщепление главных структурных компонентов соединительной ткани (эластина и коллагена) дестабилизирует ее основное вещество, нарушает межклеточные контакты и ведет к повреждению сосудов. Возникают кровоизлияния и, как следствие, некроз тканей, который, расширяя зону бактериальной инвазии, создает условия для гиперпродукции токсинов и общей интоксикации организма.
Широкий спектр экстрацеллюлярных ферментов синегнойной палочки включает два липолитических фермента — липазу и фосфолипазу С. Действуя вместе, они вызывают гемолиз — выделение провоспалительных медиаторов из тромбоцитов (лейкотриен B4 и другие эйкозаноиды), нейтрофилов (оксиданты, гидролазы) и базофилов (гистамин). Это усиливает и без того немалую флогогенность возбудителя.
Важнейший из токсинов синегнойной палочки — экзотоксин А. Механизм его действия аналогичен дифтерийному токсину (см. «Возбудитель дифтерии»). Он состоит из трех доменов, один из которых фиксируется альфа-2-макроглобулиновыми рецепторами клеток, второй обеспечивает транслокацию через плазматическую мембрану, а третий ингибирует синтез белка, блокируя рост пептидной цепи на рибосомах за счет инактивации (АДФ-рибозилирования) фактора элонгации-2 (EF-2). В активации токсина и мембранных рецепторов участвует фурин — представитель семейства клеточных протеиназ, физиологическим назначением которых является функционально значимая модификация (ограниченный протеолиз) белков-предшественников эукариотических клеток. При внутривенном введении животным (для мышей LD50 составляет 60—80 нг) токсин А вызывает поражение различных органов (гепатоцеллюлярный некроз, кровоизлияния в легкие, некротические изменения в почках), а при введении в кожу индуцирует эритематозный отек, который некротизируется в течение 2—3 дней. Это позволяет думать об участии токсина А в развитии инфекционного процесса, хотя признание за ним главных патогенетических функций сталкивается с противоречиями: 1) анатоксин, изготовленный на основе токсина А, не обеспечивает надежной защиты животных от синегнойной инфекции; 2) не все госпитальные штаммы синегнойной палочки секретируют токсин А; 3) антитоксическая терапия не дает убедительных результатов.
Синегнойная палочка выделяет два токсина, ExoS и ExoU, которые достигают клеток-мишеней через секрецию типа III. В этом случае белки непосредственно инъецируются при плотных адгезивных контактах из цитоплазмы бактерий в цитоплазму клетки; в свободном виде те же токсины неактивны. Обладая активностью АДФ-рибозилтрансфераз, ExoS реагирует с ГТФ-связывающими белками, которые контролируют передачу сигналов, участвующих в синтезе цитокинов и хемокинов. Это вызывает повышенную секрецию флогогенных медиаторов в организме, зараженном синегнойной палочкой. Полагают, что главной мишенью для токсина S являются нейтрофилы, которые в числе прочих эффектов лишаются способности мигрировать в зону инфекции. ExoU изучен меньше, но ясно, что он действует в основном на макрофаги. Каким образом токсин U убивает макрофаги, остается невыясненным. В целом, по сравнению с экзотоксином А, ExoS и ExoU обладают меньшей болезнетворностью и продуцируются ограниченным числом штаммов. К тому же максимум образования токсина S происходит не при 37°С, а при 30—32°С.
Экспрессия большинства (если не всех) факторов вирулентности неконститутивна, т.е. зависит от условий обитания и меняется в различные фазы роста. Например, протеазы лучше всего продуцируются в логарифмической и ранней стационарной фазах, когда плотность бактерий особенно высока. Этот феномен, в основе которого лежит регуляция экспрессии генов через систему аутотрансмиттеров, получил название «чувство кворума» — англ. quorum sensing.
Как и у всех грамотрицательных бактерий, в наружной мембране синегнойной палочки содержится липополисахарид, обладающий свойствами эндотоксина. При массивном поступлении в кровь он включается в развитие септического синдрома, которым нередко осложняется генерализованная синегнойная инфекция. В этом отношении синегнойная палочка повторяет другие грамотрицательные бактерии (прежде всего энтеробактерии), которые в современной клинике служат главной причиной сепсиса. Безусловно, эффект эндотоксина следует воспринимать не изолированно, а в сумме с другими факторами, тем более, что токсичность псевдомонадных липополисахаридов ниже, чем у других бактерий. Необходима обширная зона поражения (например, инфицированные ожоги), чтобы реакция на всасывание токсичных начал обрела характер каскадного септического процесса.
Мы прокомментировали два главных патогенетических механизма синегнойной инфекции — инвазию и интоксикацию. Это то, что определяет клинику, решая судьбу больного. Следует, однако, помнить, что реализация патогенетического потенциала исходно зависит от закрепления возбудителя во входных воротах инфекции. Синегнойная палочка не колонизирует здоровую кожу и слизистые оболочки, но при их физической деструкции или сопутствующей патологии такая возможность появляется. Синегнойная палочка обладает пилезависимыми и пиленезависимыми адгезинами. Они связываются с гликопротеинами соединительнотканного матрикса, которые обнажаются при повреждениях. Адгезию на слизистых оболочках обеспечивают пили и капсулоподобные экзополисахариды, обладающие сродством к муцинам, глико- и фосфолипидам мукоидного тракта.
Образующаяся биопленка вместе с нарушением мукоцилиарного транспорта объясняет высокий риск синегнойной инфекции респираторных органов при застойных явлениях в легких и муковисцидозе. От таких больных выделяются мукоидные штаммы, в изобилии секретирующие слизь. Иначе выглядит колонизация искусственных материалов: в этом случае преобладают немукоидные штаммы, которые, обладая более высокой гидрофобностью, лучше прилипают к пластиковой поверхности.
Следует отметить еще один факт. Как строгий аэроб, синегнойная палочка сильно зависит от железа, входящего в состав цитохромоксидаз. В конкуренции за ионы железа она использует различные механизмы, главным из которых является связывание и трансмембранный транспорт железа специальными белками (сидерофорами) — пиовердином и пиохелином. В организме они (особенно пиовердин) конкурируют с железосвязывающими белками хозяина — ферритином, трансферрином, лактоферрином и гемоглобином. О значении этого «соревнования» для развития синегнойной инфекции говорят наблюдения над болезнетворностью мутантных штаммов, дефектных по пиовердину. В отличие от диких двойников, они не опасны при заражении мышей в участки обожженной кожи, но обретают патогенность при введении с пиовердином.
Патология
В целом вирулентность синегнойной палочки определяется суммой факторов, которые неодинаково активны в разных фазах и при различных формах инфекционного процесса. И все-таки, в отличие от многих возбудителей пиогенной оппортунистической инфекции у нее просматриваются главные патогенетические атрибуты: мукоидное вещество (адгезия, резистентность), протеиназы (инвазия), экзотоксин А (интоксикация).
Синегнойная палочка способна поражать практически любую ткань, а связанные с ней процессы отличаются особым упорством и неблагоприятным прогнозом. Это объясняется по меньшей мере двумя причинами: 1) синегнойная инфекция обычно возникает на скомпрометированном иммунологическом фоне, при тяжелых (порой необратимых) повреждениях местных защитных барьеров; 2) возбудитель обладает высокой резистентностью к антибактериальным препаратам. Подобно всем гнойно-воспалительным заболеваниям, синегнойная инфекция может протекать в двух взаимосвязанных формах — местные процессы и генерализованная инфекция. В первом случае речь идет о калейдоскопе очаговых поражений отдельных органов, во втором — о сепсисе, т.е. о патогенетически значимой бактериемии и/или токсинемии. Для синегнойного сепсиса характерна септикопиемия, типичным проявлением которой служат вторичные (метастатические) поражения кожи эритематозно-некротического характера — ectyma gangrenosa.
Как возбудитель внутрибольничных инфекций синегнойная палочка особенно опасна в ожоговых отделениях и стационарах хирургического профиля. Это легко понять, так как именно здесь концентрируются больные с открытыми раневыми поверхностями, производятся инвазивные диагностические и лечебные процедуры, которые создают опасность внедрения бактерий в ткани и кровь в обход непроницаемого для синегнойной палочки барьера — неповрежденных слизистых оболочек и кожи. По той же причине синегнойная инфекция сопутствует поражениям роговицы (например, кератит от применения контактных линз, глазных капель), а также осложняет офтальмологические операции вплоть до панофтальмита. Аналогичный принцип срабатывает в урологических клиниках, когда синегнойная палочка попадает в мочевыводящую систему при введении нестерильных катетеров и промывающих растворов.
Чувствительность к синегнойной инфекции повышается на фоне нейтропении, которая возникает при интенсивной антиметаболитной и лучевой терапии (больные раком, злокачественными заболеваниями крови). К группам риска относятся больные диабетом, трофическими язвами, муковисцидозом. В последнем случае мукоидные штаммы синегнойной палочки колонизируют респираторный тракт и, вытесняя другие бактерии (не без помощи интенсивной антибактериальной терапии), становятся единственной причиной хронической бронхопневмонии. Бактерии размножаются внутри толстого слоя слизи (биопленка!), который образуется из муцина, в избытке секретируемого клетками респираторного эпителия, и мукоидного вещества самой синегнойной палочки. Слизь защищает ее от антимикробных факторов и антибиотиков, но не препятствует медленной деструкции слизистой оболочки протеолитическими ферментами бактерий и фагоцитов. Процесс никогда не переходит в сепсис, что, по-видимому, связано с изобилием циркулирующих антител к синегнойной палочке. Больной страдает и в конце концов гибнет от угасающей функции легких на фоне прогрессирующего фиброза.
Иммунитет и этиотропная терапия
Исходя из патогенеза синегнойной инфекции, полноценный иммунитет к синегнойной палочке должен включать два механизма — антиинвазивную и антитоксическую защиту. Антитоксический иммунитет связан с антителами, нейтрализующими токсины, прежде всего токсин А. Антиинвазивный иммунитет базируется на нейтрофилзависимом воспалении, которое определяет патогенетическую сущность гнойного процесса. В очаге воспаления нейтрофилы взаимодействуют с опсонинами (комплемент, антитела), создавая кооперацию, от которой зависит эффективность воспалительной реакции, т.е. ее способность элиминировать биоагрессивные начала, вызвавшие и поддерживающие воспаление. Потенциал иммунных реакций необходимо соизмерять с состоянием колонизационной резистентности тканей, так как ее нарушения являются обязательным условием для развития оппортунистических инфекций.
Стремление к иммунологическому управлению синегнойной инфекцией связано с повышенной устойчивостью Ps. aeruginosa к антибиотикам, которые часто оказываются неэффективными. Для иммунотерапии с большим или меньшим успехом применялись сывороточные препараты животных и человека, обогащенные антителами против синегнойной палочки (плазма, сыворотка, иммуноглобулины); в ряде случаев акцент делался на антителах, нейтрализующих токсин А. С превентивной целью испытывались вакцинные препараты, направленные на активную иммунопрофилактику синегнойной инфекции в группах риска (например, перед операциями). Для этого использовались убитые вакцины из цельных клеток, липополисахаридные эндотоксины, полисахарид внеклеточной слизи и пр. Есть данные об эффективности такого подхода, настроенного на опережающее усиление опсонического звена фагоцитарных реакций. Однако токсичность препаратов, антигенная неоднородность синегнойной палочки, гипореактивность больных людей, организационные трудности (требуется несколько инъекций) тормозят развитие данного направления. К тому же, по-видимому, не все поствакцинальные антитела обладают опсонической (а, следовательно, и антиинвазивной) активностью. Об этом говорит эффекторная неполноценность антител против синегнойной палочки при хронической синегнойной инфекции. Например, в крови больных муковисцидозом содержится множество антител против антигенов синегнойной палочки, но они не обеспечивают должного усиления фагоцитоза. В целом результаты специфической иммунокоррекции синегнойной инфекции оставляют желать лучшего. Оптимизм многих авторских начинаний оказался иллюзорным, не выдержав реалий клинической практики. За исключением скромного производственного выпуска антитоксической плазмы (специфичность ее не очень надежного лечебного эффекта остается под вопросом), сегодня нет ни одного официнального иммунопрепарата для борьбы с синегнойной инфекцией.
Противоречивые результаты получены и при лечении специфическими бактериофагами. Это отражает принципиальную бесплодность попыток создания стандартных фаговых препаратов с видовым диапазоном бактерицидности. Спектр действия каждого фага ограничен узким набором штаммов, что исключает видовую универсальность фаготропного эффекта даже в тех случаях, когда препарат комбинируется из нескольких фагов. Более логичным выглядит применение аутофагов, адаптированных к аутоштаммам, изолируемым от конкретных больных. Однако и в этом случае логика сталкивается с эволюционно закрепленной изобретательностью микробов. Бактериальные популяции неоднородны, и внутри каждой их них присутствуют (или формируются) клоны, устойчивые к селекционированным фагам. Этим, а также невозможностью обеспечить эффективный доступ фагов к микробным мишеням в очаге инвазии можно объяснить неудачи данного формально логичного принципа воздействия на синегнойную инфекцию, как, впрочем, и на другие бактерии.
Главным рычагом воздействия на синегнойную инфекцию остается антибиотикотерапия. В последние годы благодаря внедрению в клиническую практику новых препаратов здесь просматривается очевидный прогресс. Отметим, в частности, имипенем и усовершенствованные цефалоспорины типа цефоперазона, которые обладают широким спектром антипсевдомонадной активности и неплохо зарекомендовали себя в лечении синегнойной инфекции разной локализации. Впрочем, к имипенему уже имеются резистентные клоны. Они связаны с формированием мутантов по белку OprD, который выступает в роли порина, обеспечивающего проникновение имипенема в периплазматическое пространство.